
記住我




To explore the impact of reference bias on the quantification of gene expression in wheat, we simulated 1000 read pairs from each high-confidence (HC) gene in Chinese Spring RefSeq v1.1 and the nine chromosome-level genome assemblies generated as part of the 10+ wheat genomes project [20, 22] if the longest transcript of the gene is at least 500 bp. These reads were pseudoaligned or aligned to the Chinese Spring reference transcriptome or genome using kallisto or STAR, respectively. These algorithms represent pseudoalignment and alignment-based methods and are among the most commonly used tools for RNA-seq quantification in the wheat community.
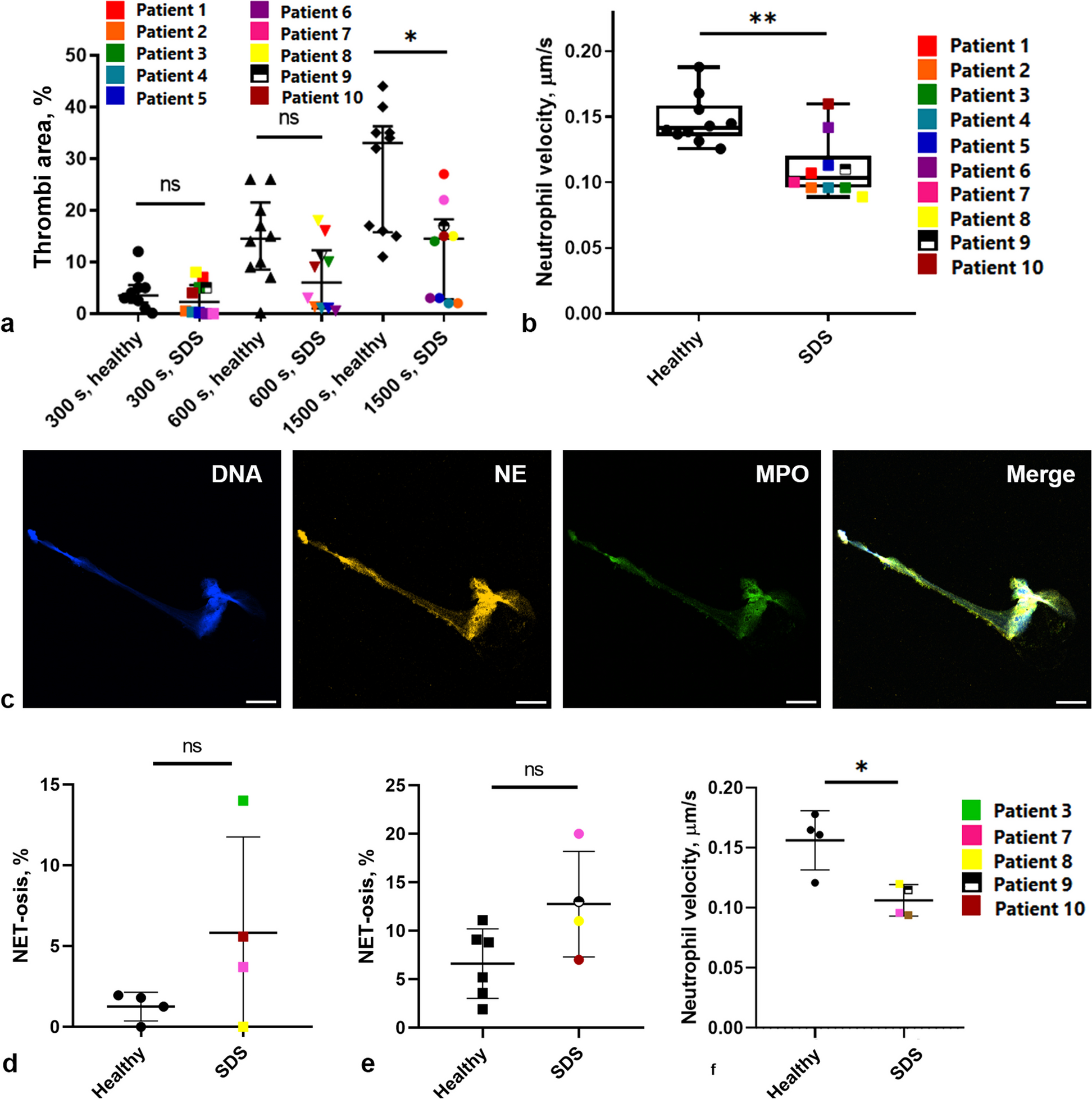
Mapping Chinese Spring reads to Chinese Spring, hereafter referred to as self-mapping, yields very accurate estimates of gene expression, with kallisto slightly outperforming STAR (Fig. 1a, b, Additional file 1: Table S1). Using kallisto, 88,401/88,443 (99.95%) of genes were correctly quantified (between 500 and 1500 read pairs). Thirty-two genes were underestimated (< 500 read pairs) and 10 genes were overestimated (> 1500 read pairs). Using STAR, 87,689/88,443 (99.15%) were correctly quantified with 504 and 250 genes underestimated and overestimated, respectively.
Fig. 1
Assessing the extent of reference bias in wheat. A Distribution of read counts when self-mapping Chinese Spring simulated reads or cross-mapping Landmark simulated reads. Comparing STAR and kallisto using the Chinese Spring RefSeq v1.0 reference and RefSeq v1.1 transcriptome and kallisto using the pantranscriptome reference. B Percentage of genes with expression estimated correctly, expression underestimated (< 500 read pairs) and expression overestimated (> 1500 read pairs) for simulated reads from 10 cultivars aligned to Chinese Spring with kallisto and STAR or to the pantranscriptome reference with kallisto. C Balance of homoeologue expression across triads when self-mapping Chinese Spring or cross-mapping Landmark simulated reads, comparing STAR and kallisto using the Chinese Spring RefSeq v1.0 reference and RefSeq v1.1 transcriptome and kallisto using the pantranscriptome reference. Each point on the ternary plot represents one triad. Points towards a corner indicate dominant expression of that homoeologue, and points opposite a corner indicate suppression of that homoeologue. D Percentage of triads in each expression category, using simulated reads from 10 cultivars aligned to Chinese Spring with kallisto and STAR or to the pantranscriptome reference with kallisto
Mapping reads generated from the other cultivars to Chinese Spring, hereafter called cross-mapping, yielded much less accurate estimation of gene expression with a skew towards underestimation (Fig. 1a, b, Additional file 1: Table S1). The percentage of genes correctly quantified ranged from 55,773/63,001 (88.53%) for Lancer, with 5700 (9.05%) and 1528 (2.43%) under- and overestimated, respectively, to 58,468/64,077 (91.2%) for Norin61, with 2527 (3.94%) and 3082 (4.81%) genes under and overestimated, respectively. For cross-mapping, unlike self-mapping, STAR appears to perform better than kallisto; the proportion of correctly quantified genes ranged from 58,390/63,001 (92.68%) for Lancer, with 3916 and 695 under and overestimated, respectively, to 59,648/64,077 (93.1%) for Norin61, with 2450 (3.82%) and 1979 (3.09%) genes under and overestimated, respectively.
To explore the effect of reference bias on the quantification of homoeologue expression balance, we calculated the proportion of triads belonging to each category that defines a different state of relative homoeologue expression. As reads were simulated evenly across genes, all triads should be classified as balanced; therefore, triads classified as imbalanced (one or two homoeologues with expression greater than the other(s)) are considered incorrectly classified. The percentage of correctly classified triads varies between 80.97% (Lancer) and 93.84% (Norin61) using kallisto and between 90.23% (Lancer) and 96.12% (Norin61) using STAR (Fig. 1c, d, Additional file 1: Table S2). Across the cultivars, triads incorrectly classified as suppressed, where one homoeologue is estimated to be expressed less than the others, were far more common than triads incorrectly classified as dominant, where one homoeologue is estimated to be expressed more highly than the others (Fig. 1d, Additional file 1: Table S2). This reflects how the reference bias leads to more underestimated than overestimated genes.
The B subgenome has the most, and the D subgenome the fewest, number of triads incorrectly classified as suppressed. This is in line with observations of greater diversity in the A and B subgenomes, with the B subgenome having the highest [16]. This difference is largely caused by gene flow from wild tetraploid T. dicoccoides to T. aestivum during the history of its cultivation, without comparable gene flow to the D subgenome [17, 19, 23]. This finding suggests the historic gene flow from tetraploid wheat likely contributes to reference bias in RNA-seq analyses.
To explore the extent of errors when comparing two cultivars mapped to a common reference, we compared the estimated expression of Lancer and Jagger genes, whose simulated reads were both aligned to Chinese Spring using STAR (Fig. 2a, b). Genes with read counts > 1.5 × or < 1/1.5 × compared to the other cultivar were classified as incorrectly quantified. Using STAR, 4791/60,338 (7.94%) genes were incorrectly quantified between the two cultivars; of these genes, 2747 and 2044 genes had a lower read count in Lancer and Jagger, respectively.
Fig. 2
The impact of reference bias on expression differences between cultivars and enrichment of incorrectly quantified genes within introgressions. A The distribution of incorrectly quantified genes in 5-Mbp windows, coloured by the cultivar in which the estimated expression is lower; orange blocks are underestimated in Lancer compared to Jagger, while green blocks are underestimated in Jagger compared to Lancer. The reads are aligned using STAR as this outperformed kallisto for cross-mapping. B Expression counts for Lancer-Jagger orthologue pairs. Genes are considered incorrectly quantified if their estimated read count is 1.5 × or 1/1.5 × the other cultivar. C CDS nucleotide identity between Lancer and Jagger 1-to-1 orthologue pairs, binned into 5-Mbp genomic windows based on Chinese Spring RefSeq v1.0. D Percentage of genes incorrectly quantified and correctly quantified in characterised introgressed regions and regions not characterised as introgressed. E CDS nucleotide identity between Lancer and Jagger 1-to-1 orthologue pairs for those that are incorrectly quantified and those that are correctly quantified. F Percentage of genes incorrectly quantified and correctly quantified, split into bins of different levels of CDS nucleotide identity
We observed a clear overlap between clusters of incorrectly quantified genes and regions of divergence between the cultivars (Fig. 2a, c), identified by blocks of reduced CDS nucleotide identity between pairs of orthologues between Lancer and Jagger. Such gene-level divergence is indicative of introgressed material; indeed, several of these blocks correspond to previously characterised introgressions. These introgressions include (coordinates based on Chinese Spring RefSeq v1.0) the following: Aegilops ventricosa introgression in Jagger (chr2A:1–24,643,290) [20, 21, 24]; Triticum timopheevii introgression in Lancer (chr2B:89,506,326–756157100) [20, 21]; Aegilops comosa introgression in Jagger (chr2D:570,141,481–613325841) [21]; and a Thinopyrum ponticum introgression in Lancer (chr3D:591,971,000–615552423) [20, 21]. 1881/3054 (61.59%) of introgressed genes (those belonging to one of the four previously characterised introgressions listed above) were incorrectly quantified between the two cultivars, compared to 2910/57,284 (5.08%) non-introgressed genes incorrectly quantified (Fig. 2d; chi-squared p-value < 2.2e − 16). Genes with an introgressed copy in Lancer tend to be underestimated in Lancer and genes with an introgressed copy in Jagger tend to be underestimated in Jagger.
In further support of CDS divergence being a predominant contributing factor to incorrect quantification, we found that incorrectly quantified genes have a mean CDS identity between orthologue pairs of 97.3% compared to a mean of 99.9% for genes correctly quantified (Fig. 2e; p-value < 2.2e − 16; 95% confidence interval ranges from 2.45 to 2.63). The percentage of genes incorrectly quantified ranges from 83.2% for genes with < 96% CDS identity between orthologues to just 2.9% for genes with ≥ 99% identity between orthologues (Fig. 2f).
Reducing reference bias by constructing a pantranscriptome referenceThe 10+ wheat genomes project generated chromosome-level de novo assembled genomes for nine wheat cultivars in addition to the reference cultivar Chinese Spring [20]. These include numerous introgressions that are the predominant source of reference bias we observe. High-quality gene annotations for these genome assemblies have been produced [22]. We constructed a pantranscriptome reference by taking the transcripts from the 107,891 Chinese Spring HC genes and adding transcripts from the nine cultivars with a chromosome-level genome assembly generated as part of the 10+ wheat genomes project [20] if that transcript’s gene exists in a 1-to-1 relationship with a gene from Chinese Spring, based on OrthoFinder [25] orthogroup assignments. This resulted in a set of transcripts from 763,877 genes from 10 cultivars, 107,891 from Chinese Spring and a mean of 72,887 from each of the nine other cultivars (Fig. 3). A total of 80,211 Chinese Spring genes had at least one 1-to-1 orthologue in another cultivar, while 59,639 Chinese Spring genes had a 1-to-1 orthologue in all nine other cultivars (Additional file 2: Fig. S1). The pantranscriptome reference was used as the transcriptome reference for kallisto pseudoalignment. After pseudoalignment, read counts and TPMs were summed across all transcripts corresponding to a given Chinese Spring gene. Kallisto splits read counts evenly across transcripts with an identical match so redundancy of transcripts does not cause problematic multi-mapping; all transcripts corresponding to a gene can thus be added.
Fig. 3
Creation of the pantranscriptome reference and how RNA-seq reads are aligned to it
To ensure using this pantranscriptome reference does not introduce any additional mapping errors from adding redundant transcripts, we compared quantified expression counts between four difference references: Chinese Spring, the pantranscriptome reference, Chinese Spring plus the Landmark transcripts from genes in a 1-to-1 relationship with a Chinese Spring gene, and the pantranscriptome reference without the Landmark transcripts. The simulated reads from Landmark were used for pseudoalignment. Of these four references, the pantranscriptome reference performed the best, with 97.53% of genes correctly quantified. Chinese Spring plus Landmark transcripts were very similar, with 97.50% of genes correctly quantified. This demonstrates that adding redundant transcripts and summing the read counts does not introduce errors in the kallisto mapping. Using the pantranscriptome reference without Landmark transcripts resulted in a slightly lower level of correct quantification, with 96.84% correctly quantified. The difference is likely due to uniquely introgressed genes in Landmark that are not present in the other cultivars. Nevertheless, due to many introgressed genes being common between cultivars, it still performed much better than just using Chinese Spring, which had 91.43% genes correctly quantified.
Using the pantranscriptome reference instead of Chinese Spring to quantify expression from the simulated RNA-seq reads resulted in much more accurate quantification for genes that were previously underestimated when cross-mapping, removing nearly all gene counts below 1000 (Fig. 1a, b). There was little change in the number of genes overquantified when cross-mapping and little difference in the distribution of read counts when self-mapping (Fig. 1a, b). The distribution of read counts shows that for Lancer, the most error-prone cultivar, the number of genes correctly quantified increased from 58,390/63,001 (92.68%) using STAR to 61,352/63,001 (97.38%) using the pantranscriptome reference. Using the pantranscriptome reference, only 2 genes remained quantified below 500 read pairs compared to 3916 genes when using the Chinese Spring reference. The number of triads correctly assigned to the balanced expression category also greatly increased when using the pantranscriptome reference (Fig. 1d). All cross-mapped cultivars had at least 99.89% triads correctly assigned as balanced; this compares to between 80.97 and 93.84% using kallisto, and between 90.23 and 96.12% using STAR to align to Chinese Spring.
Comparing Jagger and Lancer as before, this approach reduced the number of genes incorrectly quantified in one cultivar from 4971/60,338 (7.94%) to 617 (1.02%) (Additional file 2: Fig. S2). Only 23 genes (0.0381%) remain incorrectly quantified due to underestimation in one cultivar. Almost all the remaining error in both cross-mapped read counts and incorrectly quantified genes between cultivars is due to overestimation of gene expression, likely caused by copy number variation or presence/absence variation between cultivars, as opposed to divergence between orthologous gene models.
Exploring reference bias caused by introgressions in experimentally generated RNA-seq dataSimulated RNA-seq data is unlikely to capture the complete picture of a real experiment [26]. While our simulations highlight theoretical errors, it is important to assess how reference bias impacts published findings and how using the pantranscriptome reference corrects errors in real data. We reanalysed the sequencing data generated by He et al. [11]. He et al. [11] analysed RNA-seq data from 198 diverse wheat accessions, alongside enrichment capture paired-end DNA reads, to uncover eQTLs linked with homoeologue expression bias and variation in important productivity traits. Crucially for our work, they identified a set of genes whose expression exhibited negative correlation with its homoeologue across the panel. A subset of accessions possessed lowly expressed alleles in one of the homoeologues and the presence of the lowly expressed alleles was linked to various important productivity traits. This set contains 59 genes to which we have added ELF3-D1. While ELF3-D1 did not fall into the set of very negatively correlated 59 genes, it was used as case example due to its agronomic significance. Also, it still did show a negative correlation with its B homoeologue, with this expression bias associated with agronomic traits. This set of 60 genes is hereafter referred to as genes showing lack of expression correlation.
Firstly, to identify potential introgressed regions within these accessions, we mapped the enrichment capture paired-end DNA reads to Chinese Spring RefSeq v1.0 and for each 1-Mbp genomic window, calculated the mapping coverage deviation between each line and the median for that window across the accessions (Fig. 4a). Blocks of windows with coverage deviation values significantly below 1 have few reads that have mapped in this region relative to the other accessions. This is indicative of an introgression (which introduces divergent DNA that maps less well) or a deletion. We observed more divergent material in the A and B subgenomes, which is expected based on the higher levels of gene flow to the A and B subgenomes (Fig. 4a) [17, 19, 23]. The genes showing lack of expression correlation identified by He et al. [11] are enriched in genomic windows identified as introgressed or deleted (Fig. 4b), with 78.2% of these genes in a genomic window identified as introgressed or deleted in 30 or more accessions. In the rest of the genome, only 12.3% of genes are found in a genomic window identified as introgressed or deleted in 30 or more accessions.
Fig. 4
Enrichment of genes showing a lack of expression correlation in He et al. [11] within regions of divergence. A Chromosomal distribution of the number of accessions in each 1-Mbp genomic window which had mapping coverage deviation significantly less than 1 and are thus likely to contain divergent introgressed material or be deleted. B The number of genes from the set of 60 genes showing lack of expression correlation identified by He et al. [11] that are present in genomic windows identified as introgressed or deleted in 30 or more accessions
To explore the impact of the pantranscriptome reference on estimated expression, we pseudoaligned the leaf RNA-seq data from the 198 wheat accessions to both Chinese Spring and to the pantranscriptome reference. Kallisto was used for aligning to Chinese Spring instead of STAR for consistency with the analysis by He et al. [11]. 43/60 (71.7%) of genes showing lack of expression correlation (Fig. 5a) have, in 25 or more accessions, an estimated expression less than half when mapping to Chinese Spring compared to when mapping to the pantranscriptome reference. These are likely introgressed genes whose expression is underestimated when using Chinese Spring as the reference. 6/60 (10.0%) of the genes have, in 25 or more accessions, an estimated expression more than double when mapping to Chinese Spring compared to when mapping to the pantranscriptome reference (Fig. 5a). This may arise if, when using the Chinese Spring reference, RNA-seq reads were incorrectly assigned to a gene because the correct gene is too divergent and then, when using the pantranscriptome reference, those incorrectly assigned reads now have another more appropriate gene to be assigned to, resulting in fewer reads assigned to the first gene.
Fig. 5
The impact of reference bias on the quantification of gene expression in the accessions sequenced by He et al. [11]. A Estimated expression of the 60 genes identified as showing a lack of expression correlation by He et al. [11], using either the Chinese Spring RefSeq v1.1 transcriptome or the pantranscriptome reference as targets for kallisto pseudoalignment. The dashed black line represents x = y, which is the expected value if the reference is not affecting the estimation of gene expression. An accession lying on this dashed line has this gene’s expression estimated the same when using each reference. Red dots and green dots represent accessions in which a given gene has a TPM value < 50 or > 150%, respectively, when mapping to Chinese Spring than when mapping to the pantranscriptome reference. A red star indicates that in 25 or more accessions, the gene has an estimated expression less than half when mapping to Chinese Spring compared to when mapping to the pantranscriptome reference. A green star indicates that in 25 or more accessions, the gene has an estimated expression more than double when mapping to Chinese Spring compared to when mapping to the pantranscriptome reference. B Spearman’s correlation coefficient (SCC) between homoeologue pairs where one homoeologue is in the set of genes showing a lack of expression correlation identified by He et al. [11]. SCC scores were computed between AB, AD and BD homoeologue pairs and the lowest score was used. Triads in which any of the homoeologues were not present in the RefSeq v1.0 HC gene annotation were excluded. The significance of the difference between SCC scores when using the Chinese Spring reference compared to when using the pantranscriptome reference was calculated using a two-tailed t-test with no assumption of equal variance
While this shows that using Chinese Spring as the reference leads to underestimation of many of these genes, it is important to look at the impact of this on the calculated correlation between homoeologues that led to them being classified as genes of interest by He et al. [11]. We found that the SCC score between homoeologues from this set was − 0.0990 when using the Chinese Spring reference and 0.407 using the pantranscriptome reference (Fig. 5b; p-value < 2.2e − 16; 95% confidence interval ranges from − 0.603 to − 0.410). Even though this SCC value remains lower than the mean SCC (~ 0.8) reported for the entire set of homoeologues [11], it indicates that the usage of pantranscriptome as reference increases expression correlation estimates between homoeologues compared to single reference estimates.
Several regions with poor mapping coverage (mapping coverage deviation significantly below 1) in multiple accessions overlap precisely with previously identified introgressions from cultivars assembled in the 10+ wheat genomes project [20]. One such introgression is at the end of chr1D (484,302,410–495,453,186 bp, based on RefSeq v1.0 coordinates), present unbroken in 53/198 (26.8%) accessions (Additional file 1: Table S3) and shared with cultivars Jagger and Cadenza (Fig. 6a). The precise overlap of the blocks of the reduced mapping coverage in the accessions and in Jagger and Cadenza suggests that this introgression has the same origin in all these lines, and that no recombination has taken place within the introgression since its introduction. This lack of variation in its size makes it a good candidate for the following analysis. Additionally, this region was highlighted by He et al. [11] as it contains 6 of the genes showing lack of expression correlation, including ELF3-D1, which was used as a case example due to its role in heading date [27]. He et al. [11] suggest this is a terminal deletion; however, Wittern et al. [28] identified that the terminal region, including ELF3-D1, is an introgression in Cadenza and Jagger, deriving from either Triticum timopheevii or Aegilops speltoides, based on the ELF3-D1 gene model possessing an intronic deletion shared with both of these species. We can exclude Ae. speltoides as the donor species as protein alignments between the Jagger introgression and Ae. speltoides proteins showed a median protein identity of just 91.6%. As T. timopheevii does not have a genome assembly available, we cannot confirm it is the donor; however, the mapping profile of T. timopheevii reads to the Jagger genome assembly suggest it is a likely match (Additional file 2: Fig. S3). As we cannot be certain about the donor species, we will hereafter refer to this introgression as the chr1D introgression.
Fig. 6
Introgressed genes falsely identified as being less expressed due to reference bias. A Mapping coverage deviation of DNA reads across chr1D of Jagger, Cadenza, and 5 of the accessions analysed by He et al. [11]. Each point is the coverage deviation value for a given 1-Mbp genomic window. Windows with a normalised coverage score significantly different to the median normalised coverage score for that window across the set of lines being compared are coloured red. Coverage deviation values significantly below one indicates divergent material is present or a deletion has taken place, relative to the median of the rest of the set of lines. Coverage deviation values and significance values were calculated separately for the accessions and for the cultivars Jagger and Cadenza, the latter two being compared to mapping coverage values from the other cultivars whose genomes were assembled as part of the 10+ wheat genomes project [20]. The reduced coverage at the end of chr1D, the left-hand border of which is indicated by the vertical dashed black line, is the chr1D introgression, common to 53 of the 198 accessions and Jagger and Cadenza which were assembled as part of the 10+ wheat genomes project. B Expression of the wheat gene compared to its introgressed orthologue from the chr1D introgression, using either Chinese Spring or the pantranscriptome reference as targets for kallisto pseudoalignment. Orthologue pairs with TPM < 1 in both the introgressed and the wheat copy when mapping to the pantranscriptome reference were excluded. The significance of the difference between introgressed and non-introgressed orthologues when using the Chinese Spring or the pantranscriptome reference was calculated using two-tailed t tests with no assumption of equal variance
We compared the mean expression of genes from the chr1D introgression across accessions that possess the introgression to their 1-to-1 wheat orthologue across the accessions lacking the introgression. When using the Chinese Spring reference, the introgressed genes appear to be less expressed than their wheat orthologues (p-value = 0.0224, 95% confidence interval ranges from − 8.65 to − 0.679); however, when using the pantranscriptome reference, no significant difference in expression was found between the genes (Fig. 6b, Additional file 1: Table S4; p-value = 0.980, 95% confidence interval ranges from − 4.94 to 4.82).
Earlier, using simulated data, we demonstrated that reference bias can lead to incorrect assignment of expression balance across triads. To examine this phenomenon in real data, we examined the estimated expression across triads within the chr1D introgression that are also in the set of genes showing lack of expression correlation identified by He et al. [11]. When the RNA-seq reads are pseudoaligned to Chinese Spring, in lines with the chr1D introgression, ELF3-D1 appears to be lowly expressed and the expression of ELF3-B1 appears slightly elevated compared to accessions without the chr1D introgression. However, when mapped to the pantranscriptome reference, the expression of ELF3-D1 and ELF3-B1 in accessions with the chr1D introgression appears very similar to that in accessions without the chr1D introgression (Fig. 7a, b). The CDS sequence for ELF3-D1 from the introgression shares 97.0% sequence identity with ELF3-D1 in Chinese Spring, 97.6% identity with ELF3-A1 and 97.8% identity with ELF3-B1. The high divergence of ELF3-D1 from the introgression and ELF3-D1 from Chinese Spring and the greater similarity between ELF3-D1 from the introgression with ELF3-B1 from Chinese Spring explains how most reads were unable to be assigned, yet some were incorrectly assigned to the ELF3-B1, hence the slight increase in estimated expression of ELF3-B1 when using the Chinese Spring reference. The five other genes showing lack of expression correlation within the chr1D introgression also showed reduced homoeologue imbalance using the pantranscriptome reference and expression level in line with accessions without the chr1D introgression, in which the triad does not contain an introgressed D homoeologue. Four of these genes also showed a sligh
留言 (0)