Ethical statement
All chordoma patient samples in the study were obtained from consenting patients as approved by the University Health Network Brain Tumor Biobank (REB#-18-5820). Animal experiments were performed according to guidelines from the Canadian Council for Animal Care and under protocols approved by the Animal Care Committee (ACC) of the Princess Margaret Cancer Centre (Toronto, ON, Canada), Animal Use Protocol (AUP 6396).
Cell culture
The chordoma cell lines U-CH17P, U-CH17M, U-CH17S, U-CH11R, UM-Chor1 and MUG-chor1 were generously provided by the Chordoma Foundation (www.chordomafoundation.org). The cells were grown IMDM/RPMI 1640 (4:1; Sarstedt Inc) supplemented with 10% FBS and penicillin–streptomycin-glutamine (PSG; 100 U/mL penicillin, 100 μg/mL streptomycin, 292 μg/mL L-glutamine) (Gibco, Ontario, Canada). The cells were maintained in a humidified 37 °C incubator with 5% CO2 until confluency.
Silica-bead based cell surface protein capture
Cell surface protein capture (in triplicate for each cell line) was performed as previous described [9, 19] with minor modifications. The chordoma cell lines (U-CH17P, U-CH17M, U-CH17S and U-CH11R) were washed three times with ice-cold MES-buffered saline MBS buffer (25 mM MES, 150 mM NaCl pH 6.5) and a monolayer of cells were overlaid with 20 mL of 1% colloidal silica bead (LUDOX® CL colloidal silica suspension in water) with gentle shaking for 15 min at 4 °C. Excessive beads were removed, and the plates were washed three times with MBS buffer. 20 mL of 0.1% Polyacrylic acid (PAA) in MBS buffer was added to the cells with gentle shaking for 15 min at 4 °C. PAA was removed from the plates and fresh sucrose/HEPES buffer (250 mM sucrose, 25 mM HEPES pH 7.4 and 20 mM KCL) was added. Cells were scraped from the plate and centrifuged at 300 xg for 5 min. The supernatant was discarded, and the pellet was added to fresh sucrose/HEPES buffer. Cells were lysed by 3 cycles of sonication (35% amplitude, 10 s). A discontinuous Histodenz density gradient in sucrose/HEPES was prepared at different concentrations (45%, 50%, 55% and 60%) and layered in a 13.2 mL (Beckman) ultracentrifugation tube. The samples were diluted with Histodenz to a final concentration of 20% and added on top of the Histodenz gradient. The samples were centrifuged at 100,000 xg for 2 h in a SW 41Ti rotor (Beckman). After the centrifugation, the density gradient layers were removed, and the resulting pellet was washed with sodium carbonate (pH 12) solution via rotation for 15 min at 4 °C. The beads were centrifuged at 5000 xg for 20 min in a benchtop centrifuge and the supernatant was removed. Elution buffer (400 mM NaCl, 25 mM HEPES, 1% Triton X- 100, pH 7.4) was added to the beads and rotated overnight at 4 °C to elute membrane proteins from the silica beads. The eluted proteins were precipitated with ice cold acetone overnight at − 20 °C. The samples were pelleted at 10,000 xg for 10 min and the resulting pellet was solubilized with 100 µL of lysis buffer I (50% (v/v) 2,2,2,-Trifluoroethanol (TFE) and 50% PBS).
Subcellular fractionation and protein extraction
Subcellular fractionations (performed in triplicates for each cell lines) were performed as previously described [20] with minor modifications. Chordoma cell lines (U-CH17P, U-CH17M, U-CH17S and U-CH11R) were pelleted and washed three times with PBS. Cells were homogenised in lysis buffer (50 mM Tris–HCL (pH 7.4), 5 mM MgCl2, 0.1% Triton X-100 and Protease inhibitors) and kept on ice for 10 min, and further homogenised with a loose-fitting pestle. Sucrose was added to the lysates to a final concentration of 250 mM (isotonic solution). All the subsequent steps were done at 4 °C. The lysates were centrifuged at 800 xg for 15 min in a benchtop centrifuge (Eppendorf 5430R) at 4 °C to separate the nuclear fraction. The resulting supernatant served as the source of cytosol, mitochondria and microsomes (i.e., mixed membranes). The nuclear pellet was further resuspended in 2.5 mL of cushion buffer (2 M sucrose, 50 mM Tris–HCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT) and Protease inhibitors – Roche) and overlaid on 2 mL of cushion buffer in ultra-clear open top 5 mL ultracentrifugation tube (Beckman) and centrifuged at 80,000 xg for 45 min (Beckman SW 55Ti rotor). The mitochondrial fraction was isolated from the crude lysate by centrifugation at 8000 xg for 15 min. The resulting pellet was washed with a lysis buffer and spun again to retrieve the mitochondrial fraction. The resulting supernatant was centrifuged at 150,000 xg for 1 h (Beckman SW 55Ti) to isolate the microsomal pellet (mixed membranes). The supernatant served as the cytosolic fraction. Nuclear proteins were extracted from the nuclear fraction with a lysis buffer (20 mM HEPES, 400 mM NaCl, 0.2 mM EDTA) and rotated for 30 min at 4°. The pellet was passed through 18-guage needle several times and centrifuged at 9000 xg for 10 min to isolate the soluble nuclear fraction and insoluble pellet. The resulting organelle pellets (mitochondria, nuclear and microsome) were lysed in 100 µL of lysis buffer (50% (v/v) 2,2,2,-Trifluoroethanol (TFE) and 50% PBS).
Sample preparation for shotgun proteomics
The pellets obtained from the subcellular fractions were lysed by repeated freeze–thaw cycles (5 cycles, switching between a dry ice/ethanol bath and 60 °C water bath) in lysis buffer. Samples were sonicated on an ultrasonic block sonicator for five 10 s cycles at 10 watts per tube (Hielscher VialTweeter) followed by extraction at 60 °C for 1 h. Disulphide bonds were reduced with 5 mM DTT, followed by 30 min incubation at 60 °C. Free sulfhydryl groups were alkylated by incubating the samples with 25 mM iodoacetamide in the dark for 30 min at room temperature. The samples were diluted (1:5) with 100 mM ammonium bicarbonate (pH 8.0) and 2 mM CaCl2 was added. Proteins were digested overnight with 2 µg of trypsin/Lys-C enzyme mix (Promega) at 37 °C. Peptides were desalted by C18-based solid phase extraction, then dried in a SpeedVac vacuum concentrator. Peptides were solubilized in mass spectrometry-grade water with 0.1% formic acid. Liquid chromatography was directly coupled to an Orbitrap Fusion Tribrid (Thermo Scientific) and data was acquired as previously described [24]. Raw files were searched using the MaxQuant software (version 1.5.8.3) against a Uniprot human sequence database (number of sequences 42,041) with an FDR set to 1% for positive peptide spectral matches and protein identification using a target-decoy strategy [20]. Searches were performed with maximum of two missed cleavages, oxidation of methionine residues as a variable modification, and carbamidomethylation of cysteine residues as a fixed modification. Intensity-based absolute quantification (iBAQ) and label-free quantitation (LFQ) were enabled, with match between runs function disabled due to the differences in organellar proteomes. Subsequent analyses were performed using the proteinGroups.txt file. Contaminant sequences and matching decoy were removed, and proteins identified with two or more unique peptides were carried forward. iBAQ intensities were used for protein quantitation. The data was median normalised and missing values were imputed with low values (between 1 and 1.2 log2 values).
Immunohistochemical analysis
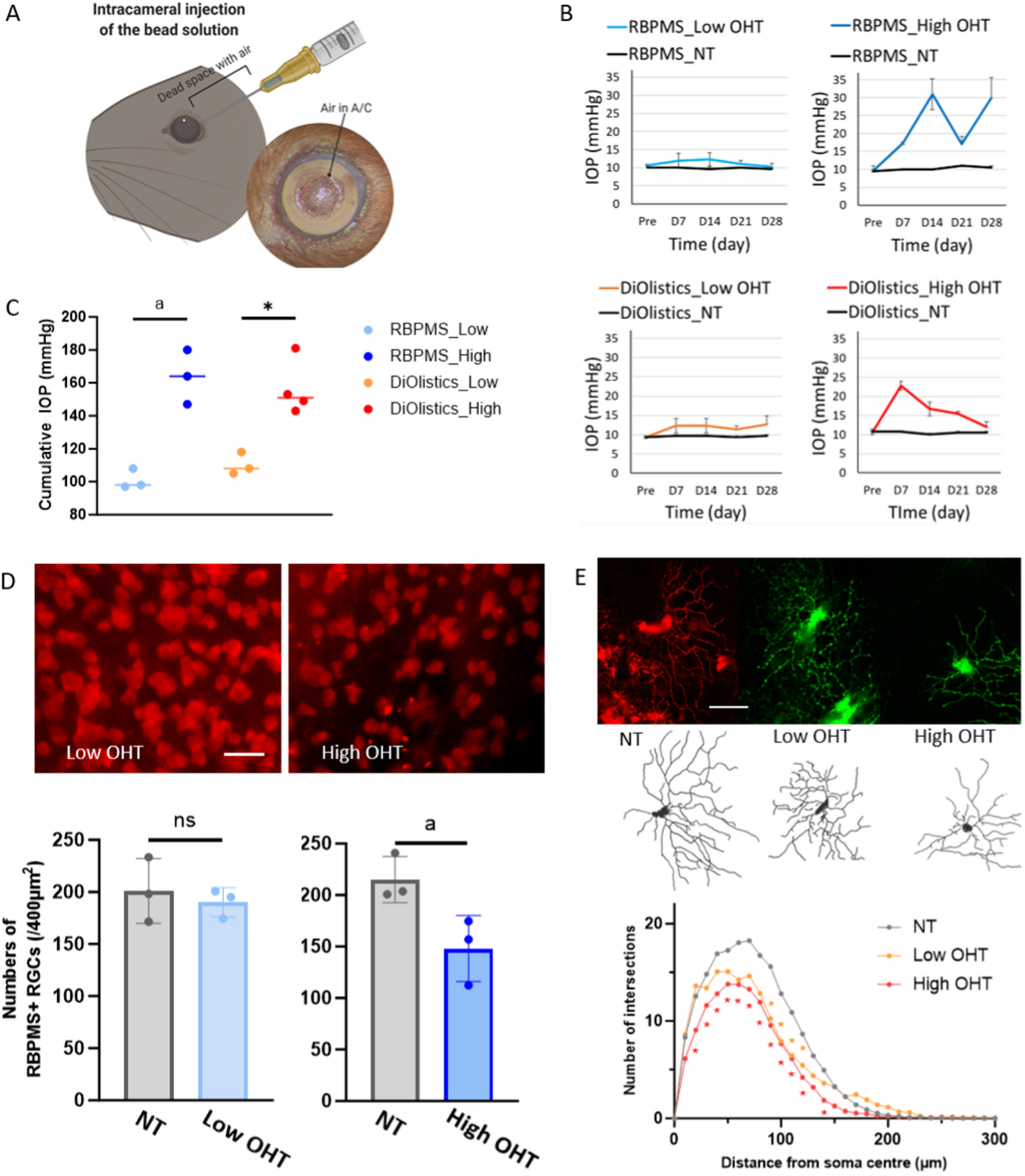
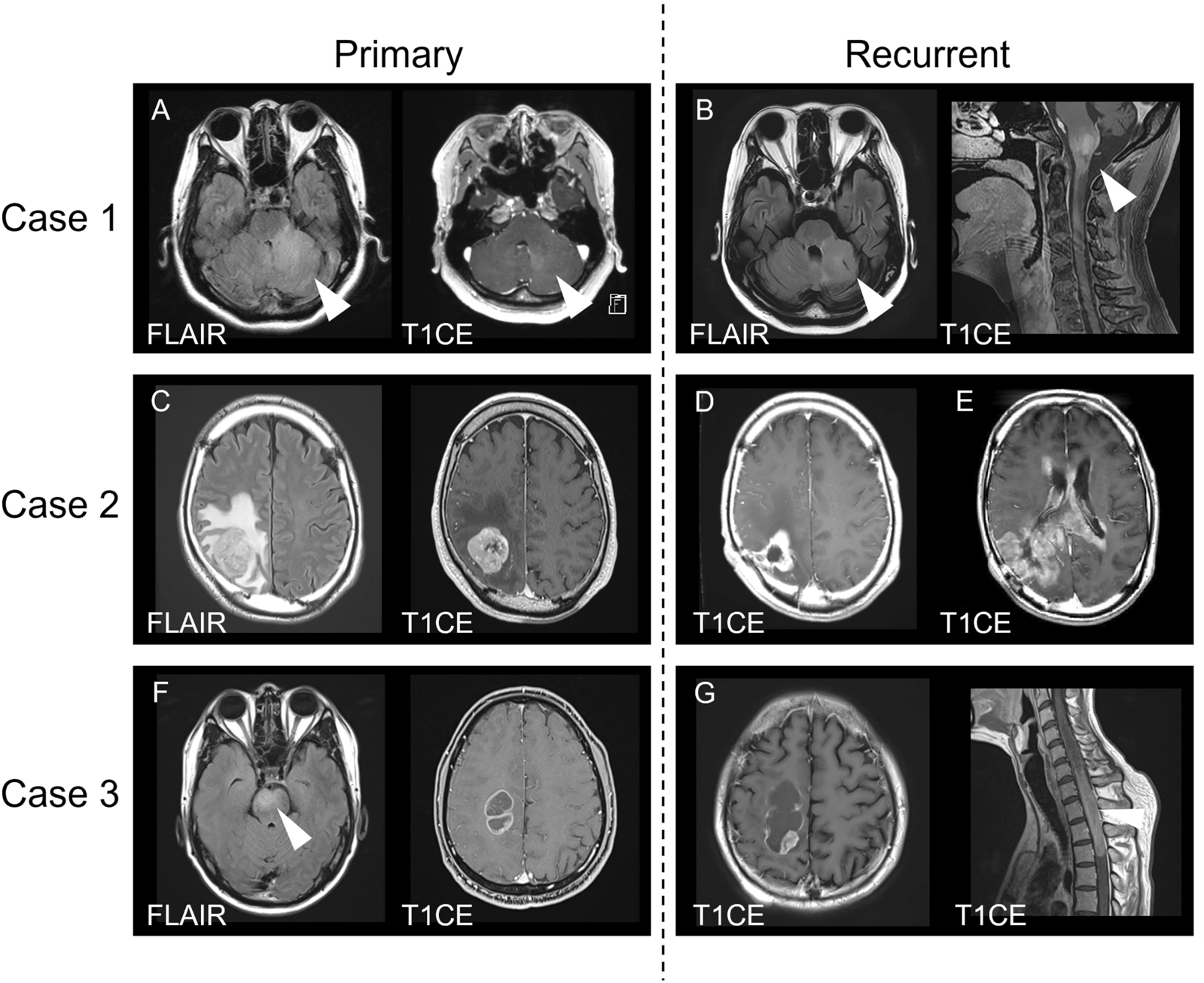
A cohort of 25 chordoma patients with formalin-fixed paraffin (FFPE) embedded slides and clinical follow up data were obtained from the University Health Network Brain Tumor Biobank (REB#-18-5820). Slides with 5 µm FFPE tissue sections were rehydrated with serial dilutions of ethanol followed by water and pH 6 sodium citrate dihydrate buffer was used for heat-mediated antigen retrieval. A 3% hydrogen peroxide in methanol solution was utilized to block endogenous peroxidase activity. Blocking solution (5% bovine serum albumin in phosphate buffered saline plus 0.1% Triton X-100) was applied to slides for 1 h at room temperature. Subsequently, primary antibody anti-PLA2R (Millipore Sigma, MABC942, mouse monoclonal antibody) were applied overnight at 4 °C diluted (1:200, 1:200, 1:250, respectively) in blocking solution. A 1 h incubation with secondary antibody was performed followed by processing with the DAKO polymer-HRP system and DAB peroxidase kit, counterstaining with hematoxylin, tissue dehydration, and slide cover slipping. Whole slide digital scanning was performed on all slides and images were analyzed using the HALO Image Analysis Platform (Indica Labs). Each slide was annotated with multiple regions of interest to delineate chordoma tumor tissue. Three independent reviewers assessed slides for all cases (JAZ, OS, and an experienced neuropathologist AG) and representative images were selected. Proportions of stain positive cells were quantified using the HALO software algorithm, defined to identify cells with either membranous or cytoplasmic staining as a fraction of all cells. This algorithm was applied to all annotated tissue sections in an unbiased systematic manner (i.e., all IHC analysis were performed in a blinded manner). Wilcoxon's rank sum test was used to compare values between skull base and spinal chordomas. Univariable and multivariable Cox models were utilized to assess the prognostic utility of stain proportions together with known major prognostic clinical factors (extent of surgical resection and radiotherapy use). For survival analyses results, the upper tertile of PLA2R1 values (samples with higher marker positivity) were compared to the lower two tertiles of values.
siRNA knockdown and clonogenic assay
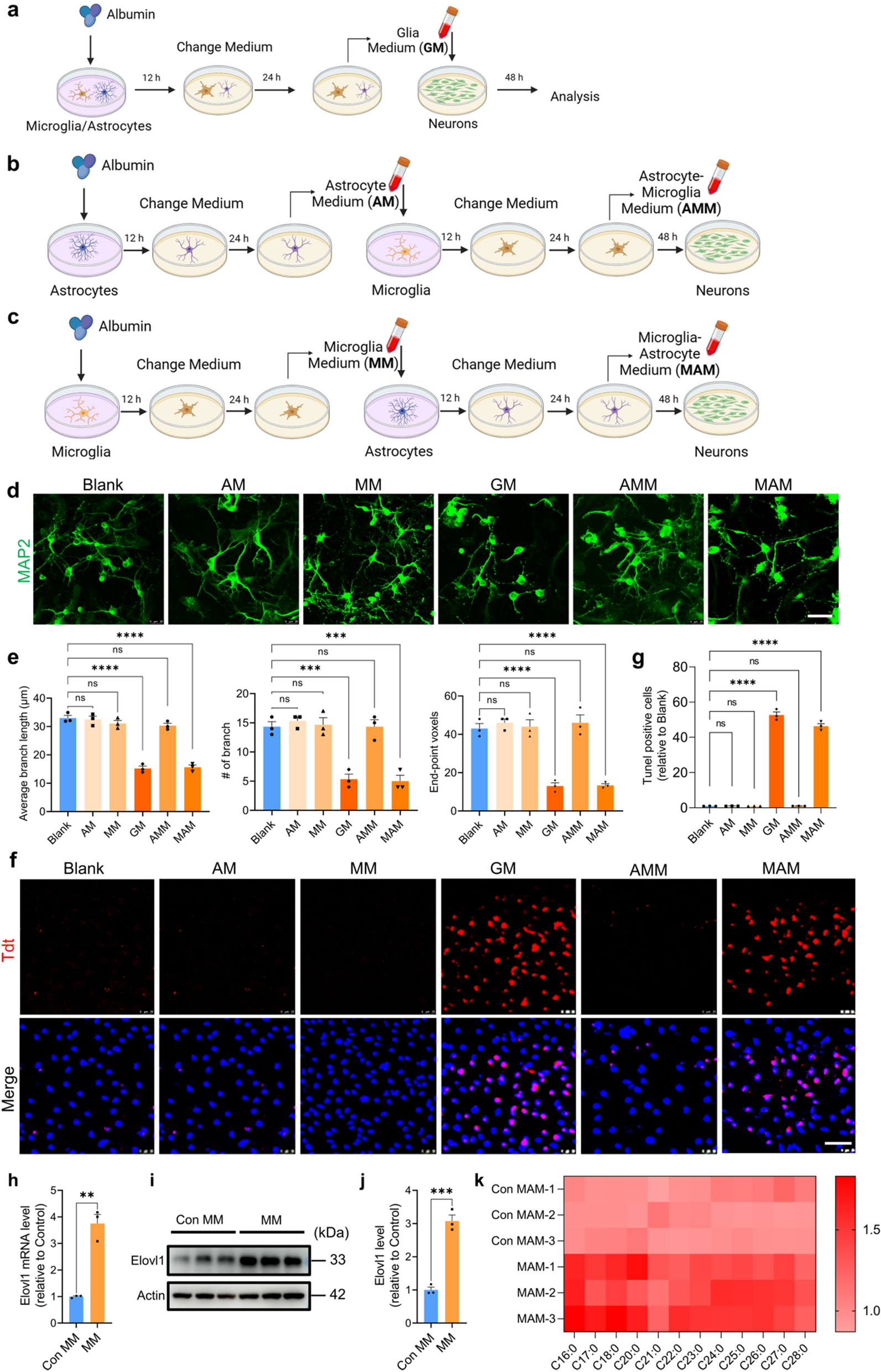
The chordoma cell lines (U-CH17M, U-CH17S and UM-Chor1) were seeded in 6-well plates at a density of 2,000 cells per well in DMEM/RPMI media and transfected with three siRNA for PLA2R1 (Cat# SR307882, Origene) and scrambled siRNA (negative control) (Cat# SR30004, Origene) at a concentration of 5 nM using lipofectamine RNAiMax transfection reagent (Invitrogen). After 2 weeks colonies were stained with crystal violet staining (0.5% crystal violet, 25% methanol) and the quantification of the colonies was performed using ImageJ (version).
PLA2R1 CRISPR/Cas9 knockdown
Two single guide RNA’s (sgRNA) targeting PLA2R1, sg1: CACCGATCACAACCTACTTCTGCAG and sg2: CACCGAGACATAACCTCATTAGCAG and control guide RNA LacZ: CACCGCCCGAATCTCTATCGTGCGG were selected from Toronto KnockOut Library V3 (TKOv3) and obtained from Integrated DNA Technologies (IDT) and were cloned into a lentiCRISPRv2 construct (Addgene) with Cas9 and Puromycin cassette. The constructs were packed into the lentivirus using 2nd generation packaging constructs. Briefly, HEK293T cells were seeded at 500,000 cells in a 6 well plate and grown in DMEM (Dulbecco's Modified Eagle Medium) (Sarstedt Inc) supplemented with 10% FBS and penicillin–streptomycin (0.1 × PS) (Gibco) for 24 h. Next, 500 ng of lentiCRISPRv2 construct (LacZ, PLA2R1 sg1 and sg2), 850 ng of psPAX2 (Addgene), and 353 ng of VSV.G (Addgene) were mixed with Xtreme Gene9 (Roche) at a 1:3 ratio (m:v) in Opti-MEM. After incubation for 30 min at room temperature, the mixture was dropwise transferred to HEK293T cells. Next day, the media was replaced with viral harvesting media (DMEM, 10% FBS, 0.2 g/ml BSA and 0.1 × PS). Viral supernatants were collected 24 h and filtered with a 0.22um filter after transfection. 10 × 105 cells UM-Chor1 cells were plated in 6-well plates containing IMDM/RPMI 1640 supplemented with 10% FBS, penicillin–streptomycin-glutamine (PSG; 100 U/mL penicillin, 100 μg/mL streptomycin, 292 μg/mL L-glutamine) and 8 μg/mL polybrene. Cells were infected with lentivirus containing lentiCRISPRv2 constructs targeting sg1-PLA2R1 (sg1), sg2-PLA2R1 (sg2) and LacZ. The UM-Chor1 cell lines without transduction (NT) were used as a negative control. After 24 h, 3 μg/mL of puromycin was added to the media for 48 h. Knockdown of PLA2R1 was verified by western blotting.
Spheroid assay
The effect of PLA2R1 downregulation on 3D growth was evaluated by seeding 10 × 103 cells CRISPR/Cas9-edited UM-Chor1 cells (sg1 and sg2) with their respective controls (NT and LacZ) in poly-HEMA-coated round-bottom 96-well plates. Cells were grown at 37 °C in 5% O2 for 24 h, and further embedded with 30ul Matrigel (Corning). The spheroids were monitored for 4 weeks, and images were obtained using a 4 × or 10 × objective on a Leica DMi1, equipped with an MC170 HD camera microscope. The quantification of spheroid size was performed using ImageJ (version 1.53 K). The metabolic activity was assessed using alamarBlue fluorometric assay. Briefly, 10ul of alamarBlue reagent was added to the spheroids and incubated at 37 °C for 24 h. The fluorescence was measured at 530 nm excitation and 595 nm emission wavelength using plate reader (Spectramax m5, Molecular devices).
Seahorse assay
UM-Chor1 negative controls (NT, LacZ) and PLA2R1 KD (sg1 and sg2) cells were plated on 96-well seahorse plate and were grown at 37 °C in 5% O2 to reach > 90% confluency. The next day, media was changed to DMEM XF assay media (Agilent) and the plate was allowed to equilibrate for 1 h in the incubator. Cartridges were prepared according to manufactures recommendations. Mitochondrial stress compounds used included Oligomycin (2 μM), FCCP (1 μM; Sigma, C2920) and antimycin A (1 μM). Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured by XF Seahorse Analyzer (Agilent Technologies).
Immunoblot
Chordoma cell line pellets and frozen chordoma tissue samples were lysed in RIPA buffer (50 mmol/L Tris pH 7.5, 150 mmol/L NaCl, 2 mmol/L EDTA pH 8.0, 0.5% (v/v) Triton X-100, and Complete protease inhibitor cocktail—Roche, Switzerland). The cells were kept on ice for complete lysis. Cell debris was removed by centrifugation at 16,000 g for 10 min at 4 °C. The protein concentration was determined by BCA assay (Thermo scientific). Commercially available normal tissue lysates from various organs (Brain cortex, cerebellum, skin, stomach, esophagus and spleen) were purchased from Takarabio (USA). Gels were loaded with 10 µg of protein lysates per lane and proteins were separated on 7, 8 or 13% SDS-PAGE gels. The resolved proteins were wet transferred to polyvinylidene fluoride membrane (PVDF) and membranes were incubated in 5% (w/v) milk in Tris-buffered saline Tween-20 (TBST; 10 mmol/L Tris-Base, 150 mmol/L NaCl, 0.05% Tween-20; pH 7.4) for 1 h. After blocking, membranes were incubated with primary antibodies (1:1000 mouse anti-human monoclonal PLA2R1, [Sigma], 1:1000 rabbit anti-human monoclonal Brachyury [Cell Signaling], 1:1000 mouse anti-human monoclonal Lamin B1 [abcam], Cleaved_CASP3 [Cell Signaling], CASP3 [Cell Signaling], cleaved-CASP7 [Cell Signaling], CASP7 [Cell Signaling], cleaved-PARP [Cell Signaling], PARP [Cell Signaling], AKT1 [Cell Signaling], AKT1 pS473 [Cell Signaling], AKT1 pT308 AKT1 pS473, FOX1 [Cell Signaling], FOX1 pT24 [Cell Signaling], GSK-3α [Cell Signaling], GSK-3α pS21 [Cell Signaling], rabbit anti-human monoclonal MEK1/2 [Cell Signaling], rabbit anti-human monoclonal MEK1/2 pS217/221 [Cell Signaling], mouse anti-human polyclonal ERK1/2 [Cell Signaling], mouse anti-human monoclonal ERK1/2 pT202/204 [Cell Signaling], rabbit anti-human monoclonal c-Myc [Cell Signaling], rabbit anti-human monoclonal c-Myc pT58 [Cell Signaling], rabbit anti-human monoclonal c-Myc pS62 [Cell Signaling, and 1:1000 rabbit anti-human monoclonal ß-actin [Novus biologicals]) overnight at 4 °C.
EdU assay
The EdU cell proliferation assay was performed using Click-IT™ Plus EdU Flow Cytometry Assay Kit (Invitrogen/Thermo Fisher Scientific). Briefly, cells were treated with 10 μM of 5-Ethynyl-2′-deoxyuridine (EdU) for 16 h at 37 °C. Cells were trypsinized and fixed with 4% PFA in PBS. Cells were permeabilized with Click-iT® saponin-based permeabilization reagent, followed by addition of 0.5 mL of Click-iT® reaction cocktail for 30 min according to manufactures instruction. DAPI (4′,6-diamidino-2-phenylindole) 0.5 μg/mL was added for 15 min. Cells were analysed using the BD LSRFortessa™ Cell Analyzer. Cells without EdU staining were used as controls.
Xenograft model
6–8 week-old male NSG mice (n = 4 per condition) were injected subcutaneously on the right or left flank with 1 × 106 of UM-Chor1 control cells (NT and LacZ) or CRISPR/Cas-9 PLA2R1 edited (sg1 and sg2) cells. Tumor growth was measured weekly using calipers as previously described [22], and tumor volume (V) was calculated with the formula V = 0.5 × l × w2, where l and w are the longest and shortest perpendicular measurements, respectively. Tumour growth was monitored for a period of 100 days after which mice were sacrificed with CO2 and tumours were resected from connective tissue and weighed.
Statistical analysis
All the proteomics experiments were performed in triplicates. Applicable data were analyzed and represented using the R statistical environment (v3.6.3). Differential expression analysis was performed using unpaired Welch's t-test for statistical analysis, and Benjamini & Hochberg adjusted p-value < 0.05 deemed as statistically significant. Visualization in R was performed using the ggplot2 (3.2.1) and ComplexHeatmap (v2.2.2).






留言 (0)