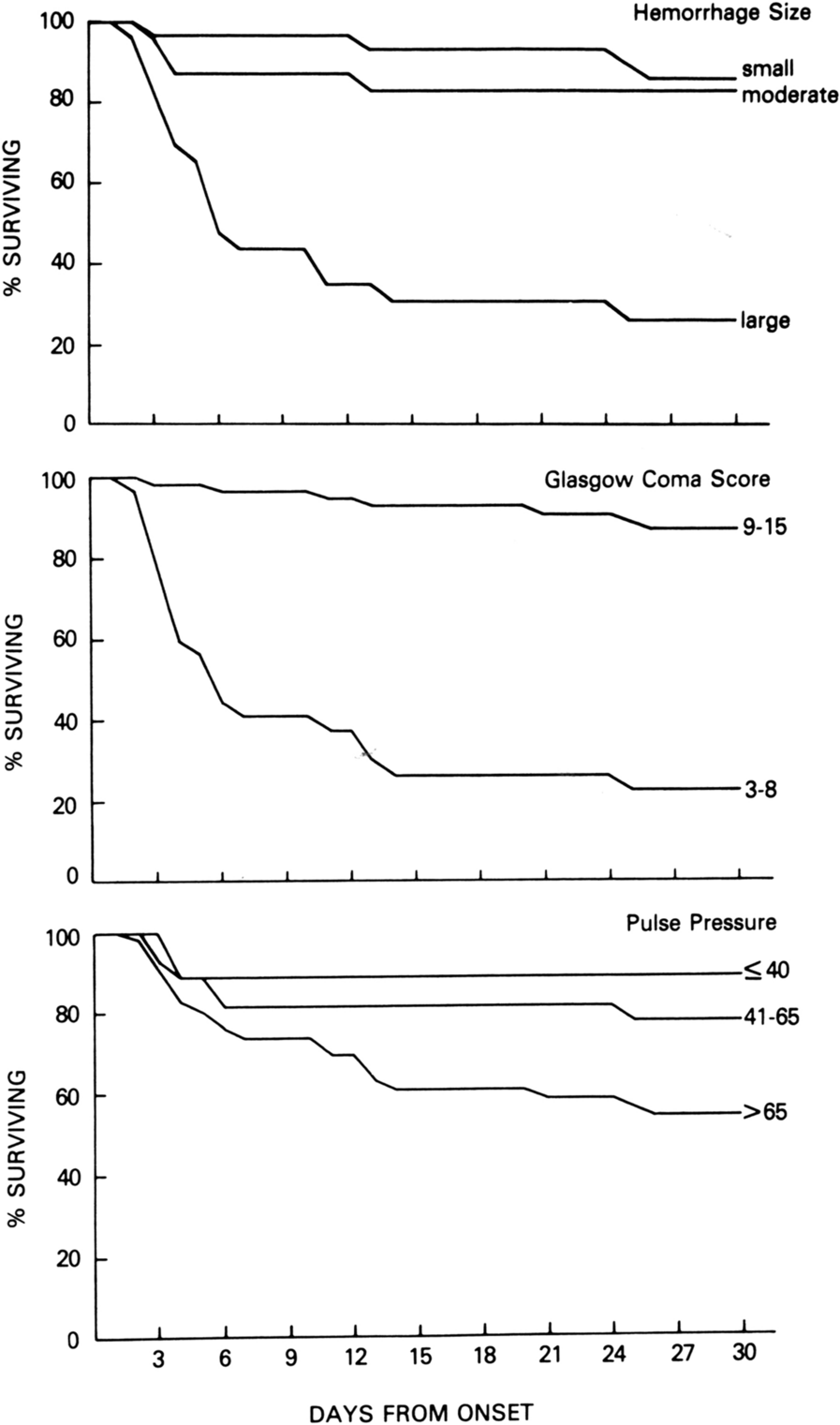
Our current in vivo murine study reports how increasing TXA dose incrementally reduces penumbral LEU mobilization after severe TBI. Furthermore, only the highest assessed TXA dose concurrently reduces penumbral BBB permeability. Additionally, TXA-treated animals consistently demonstrated enhanced Garcia Test scores, even in groups that did not benefit from reduced BBB permeability.
The benefits of TXA in correcting trauma-induced coagulopathy and improving hemostatic resuscitation are reported across multiple retrospective and observational studies. Yet, TXA appears to demonstrate impact beyond bleeding reduction. A small but similar series of reports point to TXA-related reductions in posttissue injury microvascular hyperpermeability but also occurring in sepsis and shock [12, 19, 20]. In evaluating any potential neuroprotective modulatory effects of TXA in brain injury, a clear understanding of the underpinning cellular processes occurring following TBI must first be discussed to understand any impact on TBI outcome and recovery.
Immediately following severe TBI, external mechanical forces directly deform the cerebral parenchyma, violently shifting the brain within the skull causing local tissue necrosis and microvascular disruption. These events trigger a rapid and sustained, proinflammatory immune response perpetuating local and driving more distant disruptions in homeostasis [4, 21]. Specifically, tissue hemorrhage and axonal contusions in the zone of injury stimulate damaged neuronal cells to release autocrine and paracrine signaling elements, such as danger-associated molecular patterns, which initiate a cascade that locally traffics inflammatory cells including neutrophils, astrocytes, and microglia [5, 22]. Neurovascular endothelial cells become similarly activated, lose junctional proteins and basal membrane integrity, and further enhance the proinflammatory milieu that directly leads to uninhibited extravascular fluid leakage [23].
Innate host response circulating elements are activated, and overexpress surface adhesion molecules and receptors that interact with those of endothelial cells; their interaction further promoting sustained local inflammation. These microvascular responses establish a dysfunctional and increasingly permeable BBB that perpetuates regional penumbral vasogenic edema but that may also extend beyond the local injury [23]. Based on activation and trafficking of cellular elements coupled with protease and reactive oxygen species generation, a self-sustaining inflammatory cycle deleteriously affects neuronal activity and integrity [23, 24]. Untoward consequences include neuronal cell death, oxidative stress, and axonal degeneration [4]. Unabated BBB dysfunction and consequent cerebral edema can lead to fatal herniation from elevated intracranial pressure [4].
The CRASH-2 and 3 trials revealed that TXA reduces all-cause mortality and head injury related deaths in patients with multitrauma and patients with TBI, and established the evidence base for broad implementation of TXA into prehospital and emergency department post-TBI management protocols [9, 10]. Nonetheless, it must be noted that CRASH-2 enrolled multiply-injured adults with significant bleeding while CRASH-3 enrolled adults with Glasgow Coma Scale scores 3–12 and only observed a mortality reduction with TXA in those with mild to moderate but not those with severe TBI. Although the mechanisms for this beneficial effect were initially assumed to be related to reducing postinjury bleeding as a result of antifibrinolysis, neither reduced transfusion nor reduced blood loss were observed with TXA treatment. Instead, inflammation blunting by TXA-related to dampening elements of the plasminogen activation pathway may provide a potential mechanism. As a synthetic analog of lysine, TXA competes for—and blocks—plasminogen lysine binding sites, inhibits plasmin formation and protects established clots from plasmin-mediated lysis [24]. Importantly, plasmin generation produces inflammatory cytokines, reactive oxygen species, and chemoattractants that mobilize leukocytes (monocytes and dendritic cells), and activate complement, particularly C5 and C3 [25,26,27]. Indeed, plasmin generation has been directly implicated as a driver of pervasive BBB permeability [28].
Curiously, mirroring human studies, one animal study has demonstrated how beneficial TXA effects after injury appear to be time sensitive with early administration (less than 3 h post injury) exerting protective effects, and late TXA being potentially harmful [12]. Additionally, TXA administration has also been shown to improve BBB permeability [29], particularly when given early and in male animals [12, 14]. The current study employed administration of TXA solely 1 h after TBI to specifically explore salutary effects after early administration.
With respect to TXA dosage, the CRASH trials used a 1-g bolus followed by 1-g continuous infusion over 8 h—an awkward dosage regimen based partially on extrapolated data from TXA use in menorrhagia and dental procedures in those with hemophilia [30, 31]. This difficult to manage regimen in often-chaotic operating theater conditions potentially pushed investigators to challenge and reimagine this regimen. In an attempt to challenge this, another human study, the “Prehospital TXA for TBI” trial explored doubling the initial bolus to 2 g and foregoing any subsequent infusion, discovering that in patients with intracranial hemorrhage, mortality was 30% lower if the 2 g bolus regimen was used instead of the bolus and infusion CRASH trial dosing [15]. Specifically exploring TXA dosage effects on the host immune response after severe injury, a different single-center human study, the Tranexamic Acid Mechanisms and Pharmacokinetics in Traumatic Injury (TAMPITI) trial, investigated administering boluses of placebo or 2 g and 4 g TXA in injured patients with severe bleeding [32]. They reported that higher bolus doses significantly reduced polymorphonuclear neutrophil (PMN) expression of surface L-selectin (CD62L), the primary PMN adhesion molecule responsible for LEU rolling on microvascular endothelium [32, 33]. Extrapolating these human findings to the current animal study, we sought to further explore the mechanisms behind these findings to determine whether higher doses of TXA differentially affected LEU-EC interactions in the cerebral microcirculation. The observed dose response de-escalation of live observed pial LEU rolling dovetails well with the TAMPITI findings as murine I + TXA30 is equivalent to a 2 g bolus in a 70 kg human and I + TXA60 is equivalent to a 4 g bolus. Interestingly, similarly to how only the 4 g TXA bolus reduced L-selectin expression in humans, our study found only 60 mg/kg TXA significantly reduced LEU rolling [32].
The notion that higher than CRASH-dose TXA potentially provides greater benefits after injury has also been supported by a variety of studies investigating TXA’s effects on endothelial barrier permeability. In a study by Gruen et al. [34], markers of endothelial function in the blood of patients with trauma from the Study of Tranexamic Acid During Air and Ground Prehospital Transport trial 72 h after admission were used. For every 1-g TXA increase, there was an incremental reduction in syndecan-1 plasma concentration after injury [34]. Syndecan-1 is expressed by endothelial cells and is a marker of major endothelial injury and degradation of the endothelial glycocalyx that is shed into plasma after injury [35]. Increased syndecan-1 plasma levels are identified in patients with isolated TBI, TBI with polytrauma, as well as polytrauma without TBI [36]. Of note, increased syndecan-1 concentration was associated with greater mortality in patients with TBI as well as those with polytrauma [36]. Human umbilical vein endothelial cells that are exposed to TXA demonstrate decreased glycocalyx degradation and PMN migration through the endothelial monolayer, further connecting endothelial malfunction correction to maintenance of BBB integrity by post-TBI TXA therapy [37]. The murine results presented herein align well with the above studies, demonstrating TXA treatment-driven reduced LEU-EC interactions, and better preserved neuroendothelial layer integrity 48 h post TBI. Whether higher doses of TXA may further escalate these effects remains to be determined.
Associations between post-TBI TXA treatment and improved neurological recovery were also evident in the CRASH-3 TBI groups [10]. The current animal TBI study shows that all TXA doses tested improved Garcia Test scores at both 24 and 48 h, which corroborates previous animal work that showed TXA improves neurological recovery after TBI, especially when administered less than 3 h after injury [12, 14]. This is also supported by a new murine study demonstrating significant improvements in both learning and memory in similarly injured animals exposed to TXA 1 h after severe TBI and subjected to Morris water maze exercises 7 to 14 days after injury [16]. Daglas [14] and colleagues similarly found that male mice receiving TXA after TBI exhibited both a decrease in BBB permeability and more rapid motor function recovery including recovery to a preinjury baseline. Seemingly less clear are human observational studies, for example, the recently published PATCH-Trauma trial did not note improved neuroclinical recovery, as measured by the Glasgow Outcome Scale-Extended (GOS-E) at 6 months after trauma in patients receiving prehospital TXA [38]. However, the studied population was characterized by multiorgan injury, and it is unclear what proportion of patients were accurately assessed with intracranial injury, as group demarcations only involved Abbreviated Injury Scale–head/neck or Glasgow Coma Scale scores; no information was provided regarding intracranial pathology, imaging, neurological examination, neurosurgical intervention(s), or intracranial pressure monitoring.
Another similar study evaluating prehospital TXA administration also found no improvement in GOS-E scores in patients with TBI at 6 months [15]. This study, however, included patients with moderate TBI as well as patients with penetrating head injury and did not include radiological confirmation of intracranial injury. The gross neurological scoring for mice used in our study, the Garcia test, is an acute measurement of animal neurological function, focusing on gait symmetry, balance, and reflexive reactions to touch, and difficult to consider a reliable surrogate of GOS-E which assesses intellectual and social sequelae of TBI and activities of daily living, such as one’s ability to return to work and socialize [18, 39,40,41]. In a separate study, we used the identical murine severe TBI model but evaluated mice 1 to 2 weeks later, submitting them to a Morris water maze assessment as a better analysis of cognitive recovery after murine TBI. Animals receiving TXA performed significantly better in several spatial learning and memory exercises compared to untreated counterparts [16]. Thus, taking our murine data together, cognitive improvement by TXA is apparent at 1–2 days as well as 1–2 weeks, but longer durability remains unknown.
The current inquiry presents novel mechanistic explanations for TXA-related benefits after severe TBI that are explored in a murine model. As such, there are relevant limitations that merit recognition. First, the most common human TXA administration regimen is a bolus followed by an 8-h infusion. Due to practical animal husbandry constraints, our model only used a single bolus to administer different TXA dosages. Nonetheless, as discussed above, there remains clinical controversy regarding the optimal TXA dosing approach in the wake of the established CRASH trial regimen [15, 41]. At least with respect to leukocyte-endothelial interactions, potentially higher doses than those explored in this manuscript may further blunt leukocyte mobilization to the penumbra. Also, exploring additional sham groups exposed to varying doses of TXA would have improved understanding of the putative impact of TXA on leukocyte mobilization and BBB permeability. Due to staffing and timing limitations for this project, it was thought to test multiple TXA doses in injured animals to for a quasi-dose response determination instead. Second, this was a small, murine study, and the results may not reflect how TXA functions in humans with or without TBI. Third, our study utilized only male CD1 mice. Several investigatons have revealed important sex-based differences in response to TBI and post-TBI TXA therapy, denoting that the results cannot be extrapolated to female mice [14, 42, 43]. With respect to clinical outcome, the Garcia Test score used herein (in a nonstroke model) is relatively insensitive, and animals rapidly reached a score ceiling. Although serving its basic purpose here, the Garcia Test score remains rudimentary, particularly in TBI, and needs further corroboration as can be observed with Morris water maze exercises exploring learning and memory. Finally, although both LEU rolling and in vivo BBB permeability decreased with higher dose TXA, injured hemisphere wet-to-dry ratios did not. This can at least in part, be related to pial intravital microscopy which is conducted is in a very small pial area (about 5 microns2) just within the penumbra of the injury. Such observed microscopic differences may be too small to be corroborated when the entire hemisphere (1.5 cm3) is measured for water content. If a smaller part of the injured brain could be evaluated safely (no bleeding) and reliably (the same cerebral structures, consistently) closer to the injury, perhaps wet-to-dry differences could then have been observed. Furthermore, while additional analysis identifying plasmin, TPA, and plasminogen tissue levels in the different groups would have elucidated direct mechanistic effects of TXA, this was beyond the scope of this study and will need to be assessed in future studies.



留言 (0)