
記住我


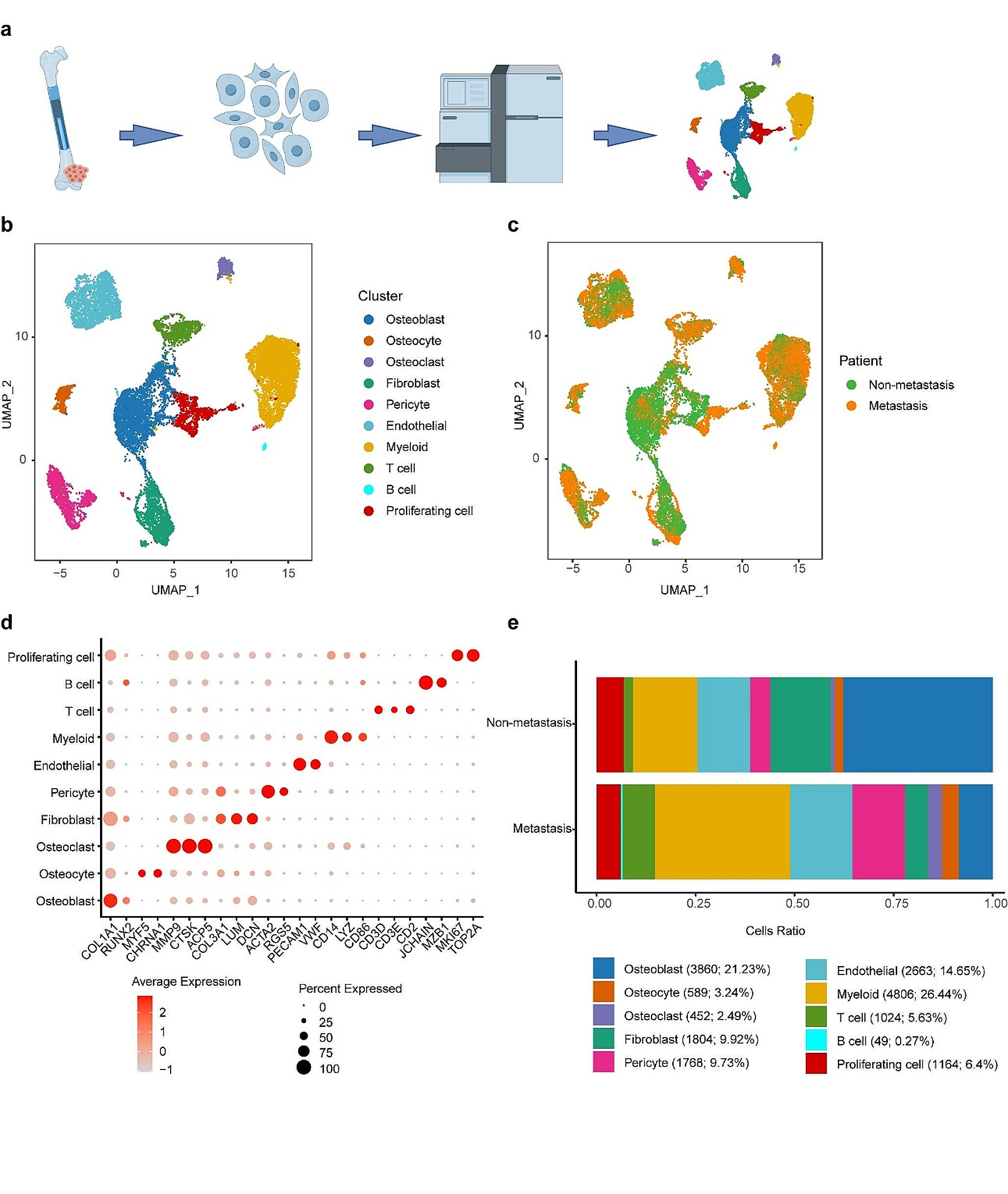




In this study, we utilized data from TCGA to calculate the expression levels of STK3 in ESCA using online software (http://ualcan.path.uab.edu/analysis.html) (Fig. 1a, b). To further elucidate the functional implications of STK3 and in ESCC, we downloaded data from the TCGA website (https://portal.gdc.cancer.gov/projects/TCGA-ESCA) and generated Kaplan-Meier curves to evaluate overall survival based on STK3 expression levels (Fig. 1c). The patients’ data achieved from our ESCC clinical database were divided into four groups according to the clinical stages (The 8th Edition of the American Joint Committee on Cancer (AJCC8) Staging Manual), and we found the IHC score difference of STK3 between early stage and the late stage of ESCC (Fig. 1d).
Fig. 1
ESCC samples showed significantly higher expression of STK3 compared to normal esophageal epithelium. (a) Bioinformatics data showed the expression difference of STK3 in cancers. (b) Bioinformatics analysis of the expression level of STK3 in ESCC, EAC, and normal esophageal squamous tissue. (c) Kaplan-Meier curve was made to evaluate the survival probability in ESCC patients according to the expression level of STK3. The data were collected from the TCGA database. (d) The patients’ data from our ESCC clinical database were divided into four groups by the clinical stages (diagnosed according to AJCC8), and the relevance was investigated. (e, f) Het-1A cells representing normal esophageal squamous epithelium, along with ESCC cell lines KYSE450, KYSE150, KYSE30, ECA109, and TE1 cells, were cultured under standard conditions; the cell lysates were subjected to Western blotting using the specific antibodies. Total RNA was extracted and subjected to Q-PCR analyses for steady-state mRNA levels of STK3. Data were presented from three independent experiments in duplicate. (g) Five pairs of ESCC samples and adjacent tissue lysates were subjected to Western blotting. (h, i) 100 pieces of ESCC samples and 80 normal esophageal epithelium were assessed via IHC. (j, k) The xenograft model of nu mice and representative image of fresh tumor tissue (n = 4 mice per group). (l) The tumor size quantification results were documented for 30 days at the 5-day interval (**p < 0.01, ****p < 0.0001.). (m) IHC analysis of tumor tissues from three groups. The tumor area was evaluated by AI assistant analysis (*p < 0.05)
Furthermore, we investigated the relative protein and mRNA expression levels of STK3 in various ESCC cell lines, including KYSE150, KYSE450, KYSE30, ECA109, and TE1, and compared to normal esophageal epithelium cell line HET-1A, by using Western blotting (Fig. 1e) and quantitative PCR (Fig. 1f), respectively. To determine the abundance of STK3 in ESCC tissue samples relative to normal esophageal epithelium tissue, we examined the protein expression levels of STK3 and p-STK3/4 using Western blotting on protein samples obtained from five pairs of ESCC tissues and normal adjacent epithelium tissues (Fig. 1g). We also investigate the expression levels of STK3 in ESCC tissue in comparison to normal tissue, utilizing IHC (Fig. 1h, i).
To investigate the impact of STK3 on ESCC tumor growth in vivo, a nude mouse model was constructed via injection of KYSE150 cells from negative control, sh-STK3#1, and STK3-OE groups. The results demonstrated that the deletion of STK3 expression substantially promotes tumor progression and augments tumor size. Conversely, overexpression of STK3 impeded tumor progression (Fig. 1j–l). The histological evaluation indicated variable levels of STK3 expression in the three experimental groups (Fig. 1m).
3.2 STK3 regulates in vitro ESCC cell proliferation, migration, and chemoresistanceIn this part, we investigated the functional role of STK3 kinase in ESCC cell lines. Specifically, we explored the effect of STK3 knockdown and overexpression on various cellular processes, including proliferation and migration. Our results demonstrated that STK3 could be activated at the Thr180 site and form hetero- or homo-dimerization with phosphor-STK3 (Thr180) or phosphor-STK4 (Thr183). To investigate the functional relevance of STK3 in ESCC, we silenced STK3 gene using shRNA loaded by lentivirus and confirmed the efficacy of knockdown through western blotting and quantitative PCR in KYSE150, TE1, and ECA109 cell lines. The CCK8 assay showed a significant reduction in proliferation rates in the knockdown group compared to the NC group (Figs. 2a, b, and S1d). EdU assay revealed that STK3 deletion led to an increase in EdU-positive cells, indicating an association between STK3 deletion and cell proliferation in KYSE150 and TE1 cell lines (Figs. 2c, d, and S1a). Furthermore, we found a positive correlation between STK3 knockdown and increased cell migration through transwell and wound scratch assays (Figs. 2e, f, and S1e, f).
Fig. 2
STK3 regulates in vitro ESCC cell proliferation, migration, and chemoresistance (a, b) KYSE150, TE1 cells stably expressing shRNA specific to STK3 were subjected to Western blotting, and quantitative PCR was shown. The CCK8 assay was presented as well. (c, d) EdU assay showed that EdU-positive cells increased according to the STK3 deletion. (e) KYSE150, TE1 cells stably expressing shRNA specific to STK3 were incubated for 36 h and then subjected to transwell assay. Migrated cells were fixed by 4% formalin and stained with crystal violet. Scale bar = 100 μm. (f) KYSE150 and TE1 cells stably expressing shRNA-STK3 were seeded in the 6-well tissue culture plates, and wound width of KYSE150 and TE1 cells with or without STK3 stably deletion at 0 and 16 h were measured. (g) KYSE150, TE1 cells stably expressing pcDNA-STK3 (STK3-Overexpression, STK3-OE) were cultured, and cell lysates were subjected to Western blotting and Q-PCR analysis. (h, i) KYSE150, TE1 cells transfected with pcDNA-STK3 were cultured and subjected to CCK8 assays and Eud assay. (j) Transwell assay showed the migration cells decreased in the STK3-OE group. (k) KYSE150 and TE1 cells with or without STK3 overexpression were seeded in 6-well plates, and wound width was measured at 0 and 36 h separately (Data are presented as the means ± standard error (S.E.). (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
To determine the effect of STK3 overexpression on ESCC, we transfected STK3-targeting pc-DNA to express high STK3 levels in three ESCC cell lines (Figs. 2g and S1g). Our results demonstrated that STK3 overexpression inhibited the proliferation and migration phenotype of ESCC cells, as confirmed by CCK8, transwell assays, and wound scratch assay (Figs. 2h, j, k, and S1h, i). EdU-positive cells decreased significantly as well (Fig. 2i). Drug resistance to cisplatin was also measured. In two cell lines, the IC50 values of the shSTK3#1 group were statistically significantly higher than those of the NC group, suggesting increased drug resistance to cisplatin (Fig. S1b). Additionally, STK3-overexpressed cells exhibited an increased sensitivity to cisplatin (Fig. S1c). In summary, our data suggest a potential inhibitory role of STK3 in ESCC proliferation, viability, and cell migration.
3.3 A moderate level of cellular ROS induces autophosphorylation of STK3, correlating with tumor suppressionAutophosphorylation of STK3 induced by H2O2 was further investigated. The findings revealed that a moderate concentration of H2O2 could elevate cellular ROS levels, consequently promoting the time-dependent upregulation of p-STK3/4 occurring within 20 h (Fig. S2a, b). Within 20 h, a concentration of 0.4 mM H2O2 was found capable of inducing autophosphorylation of STK3 in both KYSE150 and TE1 cells. Conversely, phosphor-ERK1/2, a classic phosphorylation protein known for its pro-cancer role, did not display any noticeable augmentation in the H2O2 stimuli model (Fig. 3a, b). Moreover, the concentration gradient model showed that the expression of p-STK3/4 was significantly upregulated at 0.4 mM H2O2 (Fig. 3c), and H2O2 treatment further elevated the p-STK3/4 level in STK3-OE groups (Fig. S2c). We used flow cytometry to examine the ROS level in the H2O2 concentration gradient model. Treatment with a specific concentration of H2O2 improved the FITC positive cell proportion, indicating a concentration-dependent increase of ROS level in the H2O2 concentration gradient model (Fig. S2d, f). In the rescue experiment, N-acetylcysteine (NAC) was used to eliminate intracellular ROS, and western blotting was performed to determine the expression level of phosphor-STK3/4 (Fig. 3f). The flow cytometry results showed that NAC treatment could reduce the proportion of FITC-positive cell (Fig. S2e, g). Overall, the results indicated that a moderate concentration of H2O2 treatment could induce cellular ROS accumulation and the autophosphorylation of STK3 in KYSE150 and TE1 cells.
Fig. 3
A moderate level of cellular ROS induces autophosphorylation of STK3, correlating with tumor suppression. (a, b) KYSE150 and TE1 cells were exposed to 0.4 mM H2O2 diluted in DMEM. Cell lysates were collected at 4-hour intervals and analyzed by Western blotting with specific antibodies. (c) KYSE150 and TE1 cells were treated with different concentrations of H2O2 (0–1.0 mM) for 20 h, and the cell lysates were subjected to Western blotting. (d) KYSE150 and TE1 cells were cultured for 24 h in 96-well plates, then treated with H2O2 (0, 0.2, 0.4, 0.6, 0.8, 1.0 mM) and subjected to CCK8 assays. (e) For transwell assay, cells were seeded in the 24-well chambers and treated with 0 or 0.4 mM H2O2 for 24 h. Migrated cells were fixed by 4% formalin and stained with crystal violet. Scale bar = 100 μm. (f, g) KYSE150 and TE1 cells were treated with H2O2 (0.4 mM) with or without NAC (5 mM, diluted in ddH2O) for 24 h, and the cells were subjected to the CCK8 assay. The lysates were subjected to Western blotting to evaluate the protein phosphorylation level. (h, i) The Same culture conditions for the EdU assay and the EdU-positive cells were measured between groups; analysis was shown as the means ± S.E. from three independent experiments. (j) KYSE150 and TE1 cells were treated with different concentrations of H2O2, and the cell suspension was processed and measured according to the manufacturer’s instructions. (k) KYSE150 and TE1 cells were treated with H2O2 with or without NAC, and all samples were processed according to the instructions and were subjected to the luminometer. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.)
Furthermore, cell viability was assessed at different concentrations of H2O2, revealing that 0.4 mM was a moderate concentration for ESCC cell lines’ growth, while high concentrations proved lethal to the cells (Fig. 3d). The study then investigated the migration ability of KYSE150 and TE1 at the moderate H2O2 concentration of 0.4 mM. Both cell lines exhibited a significant decrease in migration proportion in the 0.4 mM H2O2 stimulation group, consistent with the upregulation of p-STK3/4 (Fig. 3e). The NAC rescue group was also observed to efficiently reverse the suppressive effect of H2O2 on ESCC cell proliferation, as shown in the CCK8 assays (Fig. 3g) and EdU assay (Fig. 3h, i). These findings suggest that H2O2 treatment can regulate ESCC cell viability and migration ability through the induction of ROS accumulation and the autophosphorylation of STK3.
Adenosine triphosphate (ATP) is a critical supplier of phosphate groups essential for the dimerization of serine/threonine kinase. To investigate further the mechanism of STK3 autophosphorylation under H2O2-induced oxidative stress, we examined cellular ATP levels in varying H2O2 concentrations. Our findings suggest that there exists a potential correlation between cellular ROS and an increased ATP level, while higher levels of H2O2 appear to impair the activity of H+-ATP synthase, thereby resulting in a reduction in ATP levels (Fig. 3j, k) [17].
It has been reported that elevated H2O2 levels can disrupt cellular processes by triggering necrosis and apoptosis. Consequently, we have identified STK3 as a crucial kinase in the Hippo pathway, showing a significant relationship between its phosphorylation state and kinase activity. Therefore, the concentration of H2O2 mentioned earlier (0.4 mM) was deemed appropriate for modifying the function of STK3, making it a recommended protocol for other researchers.
3.4 Activated STK3 coactivate with FOXO1 and phosphorate FOXO1 at forkhead domain Ser212While previous studies have identified FOXO1/FOXO3 as downstream substrates of STK4, the function of STK3, despite its unique role as a homolog kinase, has received limited attention. Our study aimed to investigate the interaction between STK3 and FOXO1 and the potential phosphorylation site of FOXO1 by STK3. Using immunoprecipitation analysis and western blotting, we demonstrated that STK3 interacts with endogenous FOXO1 in three ESCC cell lines (Fig. 4a). To confirm this interaction, silver staining was used to detect interaction proteins of IP samples (Fig. 4b). The protein bands were observed at the target position, which is consistent with the molecular weight of the interaction protein. Overexpression of STK3 was found to enhance the interaction between STK3 and FOXO1 in KYSE150, ECA109, and TE1 cell lines (Fig. 4c). We also used online analysis software to identify several important reported phosphorylation sites (serine 212 site and serine 256 site) of FOXO1 at a different domain (Fig. S3a).
Fig. 4
Activated STK3 coactivate with FOXO1 and phosphorate FOXO1 at forkhead domain Ser212 (a) Immunoprecipitation samples and Input/IgG samples of STK3 and FOXO1 in KYSE150, ECA109, and TE1 cells, the lysates were subjected to the Western blotting. Immunoglobulin-G (IgG) was used as the negative control, and Input as a positive control. (b) The STK3 and FOXO1 immunoprecipitation samples were subjected to Western blotting. The gels were processed according to the silver staining instructions. (c) IP samples of KYSE150, ECA109, and TE1 cells with or without STK3 overexpression were subjected to Western blotting using the specific antibodies. IgG was used as the negative control, and Input as the positive control. (d) KYSE150 and TE1 cells stably expressing shRNA-STK3 were subjected to western blotting, and the p-FOXO1212 and p-FOXO1256 expressions were specially measured. (e) The total protein and mRNA expression of FOXO1 were evaluated (ns, not significant). (f) The KYSE150 cells with or without STK3 deletion were transfected with siRNA-FOXO1#1 and pVL2-CMV-FOXO1S212A. The cell lysates were subjected to western blotting using specific antibodies. (g) KYSE150 and TE1 cells stably transfected with the empty vehicle, shRNA-STK3, or pcDNA-STK3 were cultured in 6 cm plates for at least 24 h. The cytoplasm and nucleus proteins were extracted according to the manufacturer’s instructions. β-Actin and Lamin B were used respectively as the cytosolic and nuclear marker proteins to ensure the validity of our protocol. (h, i) KYSE150 and TE1 cells stably transfected with NC, shRNA-STK3 or pcDNA-STK3 were cultured in 6-well plates with or without coverslips. We collected the cell lysates and subjected them to Western blotting. We processed the coverslips according to the immunofluorescence staining protocol and then used the microscope to record the fluorescence distribution of FOXO1. (j) The KYSE150 cells with or without STK3-OE were transfected with siRNA-FOXO1#1, and pVL2-CMV-FOXO1S212A were cultured in 10 cm plates, and the cell lysates using an anti-FOXO1 antibody for immunoprecipitation
Deletion of STK3 disrupted the competitive phosphorylation of FOXO1 at the Serine212 site, leading to an elevation in p-FOXO1Ser256, a site typically phosphorylated by Akt kinase (Fig. 4d). We also examined the total FOXO1 protein and FOXO1 mRNA in the sh-STK3#1, sh-STK3#2 and NC groups. The results showed that deletion of STK3 did not affect the total protein nor mRNA expression of FOXO1 (Fig. 4e). We further identified the phosphorylation relation of STK3 kinase on FOXO1 by transfecting FOXO1 mutant plasmids of arginine to replace serine at the 212 site in KYSE150 and TE1 cell lines (STK3 deletion group and NC group separately). This revealed a significant downregulation of p-FOXO1Ser212 in FOXO1Ser212A and FOXO1 knockdown group, compared to FOXO1wt in the NC group (Figs. 4f and S3b). Thus, we successfully demonstrated that FOXO1 could interact with STK3 and be phosphorylated by STK3 at the Serine212 site. Mutation of FOXO1 Serine212 using its corresponding mutant plasmids could abrogate the level of p-FOXO1Ser212 in parallel.
Previous research demonstrated that FOXO1 and FOXO3 could translocate from the nucleus to the cytoplasm in response to Akt phosphorylation at Serine256(FOXO1)/Serine253(FOXO3) and FOXO1/FOXO3 subsequent binding with 14-3-3 protein. The competitive inhibition of the phosphorylation on Serine256(FOXO1)/Serine253(FOXO3) could reverse the shuttle [18,19,20]. In the present study, we provide evidence that FOXO1 undergoes translocation from the cytosol to the nucleus in response to the activation of STK3. In this study, we conducted nuclear and cytoplasmic extraction experiments to investigate the distribution of FOXO1 in various test groups. Our results indicate that FOXO1 translocates to the nucleus in the STK3 overexpression group with an upregulating trend of p-FOXO1Ser212, in contrast to the sh-STK3#1 group or NC group (Fig. 4g). Immunofluorescence staining was employed visually display the distribution of FOXO1. As shown in Fig. 4h, i, upregulation of STK3 resulted in the accumulation of endogenous FOXO1 in the nucleus, whereas in the STK3 deletion group, the fluorescence of FOXO1 was dispersed in the cytoplasm. We conducted a co-immunoprecipitation assay to further elucidate the mechanism. Our results demonstrate that STK3 overexpression facilitates the separation of 14-3-3 and FOXO1. We also found that invalidation of FOXO1Ser212 inhibits the phosphorylation function of STK3 kinase on FOXO1, which promotes FOXO1 combination with 14-3-3 and export to the cytoplasm (Fig. 4j and S3c). These findings suggest that STK3 plays a role in regulating the distribution of FOXO1 through its phosphorylation of FOXO1Ser212 and the non-engagement between FOXO1 and the 14-3-3 protein. Notably, the translocation of FOXO1 to the nucleus may augment its transcriptional function, while its translocation to the cytoplasm may attenuate this effect.
3.5 FOXO1 inhibits tumor progression and promotes apoptosis in ESCCThe FOXO subfamily is involved in various critical cellular processes in mammalian cells, including apoptosis, stress resistance, cell cycle arrest, DNA damage repair response, and glucose metabolism, as evidenced by previous studies [21]. To investigate the specific role of FOXO1 in ESCC cell lines, we used a specific siRNA to knock down FOXO1 expression. We then utilized western blotting and Q-PCR to assess the effects of FOXO1 knockdown on protein and gene expression (Figs. 5a and S3d). The impact of FOXO1 knockdown on ESCC cell proliferation and migration was evaluated using CCK8 and transwell assays (Figs. 5b, c and S3e).
Fig. 5
FOXO1 inhibits tumor progression and promotes apoptosis in ESCC. (a) KYSE150 and TE1 cells were transfected with siRNA-FOXO1, and protein and mRNA levels were assessed through western blotting and Q-PCR. (b, c) Cell viability and migration were evaluated by CCK8 assay and transwell assay (***p < 0.001, ****p < 0.0001.). (d) FOXO1 was overexpressed by pET28a-FOXO1 transfection in KYSE150 and TE1 cells, and cell lysates were subjected to western blotting by using specific antibodies. (e) KYSE150 and TE1 cells overexpressing FOXO1 were cultured for 48 h, and the supernatants and cells were collected and washed with PBS. The cell pellets were stained by propidium iodide and Annexin V-FITC and incubated for 15 min; then, the cell suspension was subjected to a flow cytometer for the apoptosis analysis. (f, g) KYSE150 and TE1 cells overexpressing FOXO1 were cultured for 48 h, and the cells were collected and washed with PBS, and incubated in 70% ethanol overnight. The cell lysates were washed by PBS the next day, and processed with propidium iodide stain for 30 min. The cell suspension was subjected to a flow cytometer for the cell cycle analysis. (h) In the rescue experiments, the cells expressing pcDNA-STK3 were transfected with or without siRNA specific to FOXO1. The protein and mRNA were examined by western blotting and Q-PCR. (i) In the rescue experiments, the cells transfected with the targeted gene were processed and subjected to a flow cytometer for apoptosis analysis. (j) In the rescue experiments, the cells transfected with the targeted gene were processed and subjected to a flow cytometer for cell cycle analysis. (k) The transfected cells were seeded in the 24-well chamber for the transwell assay (*p < 0.05, **p < 0.01, ***p < 0.001.)
The pET28a-FOXO1 and empty vehicle were transfected into the KYSE150 and TE1 cell lines to investigate the effects of FOXO1 amplification. The results showed that FOXO1 amplification increases the apoptosis cell in both cell lines and upregulates the downstream pro-apoptotic proteins BIM and cleaved caspase 3 (Fig. 5d, e). The cell cycle assay was also conducted, revealing an increased G0/G1 ratio in the FOXO1 overexpression group. This observation suggests that FOXO1 plays a crucial role in inducing cell cycle arrest (Fig. 5f, g). Further western blotting and Q-PCR results from rescue experiments demonstrated that the overexpression of STK3 enhanced the anti-tumor function of FOXO1, evident in increased levels of BIM and cleaved-caspase 3. The deletion of FOXO1 in STK3 overexpression cells could reverse the downstream gene expression (Fig. 5h). Overexpression of STK3 increased the proportion of early and late apoptotic cells, which could be rescued by FOXO1 knockdown (Fig. 5i). The cell cycle assay revealed a consistent outcome, wherein the G0/G1 ratio demonstrated a notable increase in the STK3-OE group. Conversely, the introduction of FOXO1 knockdown was observed to reverse the cell cycle arrest (Fig. 5j). The results of the transwell assay indicated that the decreased migration effect of STK3 overexpression on ESCC cell lines could be reversed by FOXO1 deletion (Fig. 5k).
3.6 STK3 activates the FOXO1-TP53INP1/P21 axisDespite our demonstration that FOXO1 is a substrate of STK3 kinase, the downstream effector remains unknown. To explore this, we used RNA sequencing and overlapped the commonly differentially-expression genes of STK3 kinase and transcription factor FOXO1 using an online tool (http://bioinformatics.psb.ugent.be/), and we found 132 genes among the overlapping region (Fig. 6a). After mapping all differentially expressed genes to terms in a database, we focused on the gene related to positive regulation of the cell death pathway (https://metascape.org/) (Fig. 6b). The presented findings demonstrate that the expression of TP53INP1 (Tumor protein p53-induced nuclear protein 1) and CDKN1A (Cyclin-dependent kinase inhibitor 1, also known as P21) was significantly reduced in both si-STK3 and si-FOXO1 groups, as indicated by the heatmap (Fig. 6c). According to the previous study, TP53INP1 and P21 might be the critical transcriptional downstream gene of the FOXO1 [22]. Subsequently, we identified TP53INP1 and P21 as the putative downstream effectors of STK3/FOXO1. Western blotting and quantitative PCR analyses showed a significant reduction in TP53INP1 and P21 expression levels in the FOXO1 deletion group (Fig. 6d, e), and their expression levels were substantially downregulated by FOXO1 knockdown. Conversely, the overexpression of FOXO1 resulted in a concomitant upregulation of TP53INP1 and P21 (Fig. 6f, g), which suggested that the expression of TP53INP1 and P21 are closely regulated by of FOXO1. We utilized chromatin co-immunoprecipitation with antibodies to further demonstrate the regulatory relations. The results supported the transcriptional relationship of FOXO1 and TP53INP1/P21 in ESCC cell lines (Fig. 6h, i).
Fig. 6
STK3 activates the FOXO1-TP53INP1/P21 axis. (a) Venn diagram was made according to the RNA-Seq results. (b) The top 10 Gene Ontology biological processes were obtained via online bioinformatics software, and the Pathway and Process Enrichment analysis was exhibited. (c) The overlapping part of differentially expressed genes of si-STK3 and si-FOXO1 were presented in a heatmap format. (d, e) KYSE150 and TE1 cells were transfected with siRNA specific to FOXO1, and cell lysates were subjected to western blotting and Q-PCR. (f, g) FOXO1 was overexpressed by transfecting pET28a-FOXO1 in KYSE150 and TE1 cells, and cell lysates were subjected to western blotting and Q-PCR. (h, i) The ChIP assay showed that the promoter fragments were shown as amplified DNA bands. (j, k) KYSE150 and TE1 cells were transfected with the empty vehicle, and the cells stably expressing sh-STK3 were transfected with or without si-FOXO1, and the cells transfected with si-FOXO1 or si-TP53INP1 or si-P21. All the cell lysates were immunoblotted with the indicated antibodies
The downregulation of TP53INP1 and P21 was also found to be associated with the deletion of STK3, and the double knockdown of STK3 and FOXO1 resulted in significantly reduced expression levels of TP53INP1 and P21 in KYSE150 and TE1 cell lines (Fig. 6j, k). Taken together, these results highlight the crucial role of FOXO1 in mediating the transcription of TP53INP1 and P21 in ESCC cell lines. STK3 deletion significantly weakened the FOXO1 transcriptional function in ESCC, leading to a downregulation of downstream genes.
3.7 TP53INP1 and P21 act as antitumor effectors mediated by FOXO1 in ESCC cell linesThe protein TP53INP1 is recognized as having antiproliferative and proapoptotic properties, playing a role in the cellular stress response. To investigate the impact of TP53INP1 in KYSE150 and TE1 cells, we used western blotting and Q-PCR experiments to verify the knockdown efficiency of specific siRNA-TP53INP1. We also testified the function of TP53INP1 by CCK8 assay (Fig. 7a). Transwell assay was used to measure the migration cells between the NC and si-TP53INP1 group (Fig. 7b). Additionally, we conducted flow cytometric analysis to demonstrate the pro-apoptotic function of TP53INP1 (Fig. 7c, d). As aforementioned, we performed rescue experiments to elucidate further the direct involvement of TP53INP1 in FOXO1 activation (Fig. 7e, f). Our results showed that transfecting FOXO1-specific siRNA reduced the proportion of apoptotic cells in ESCC cell lines. However, transfecting both pCMV-TP53INP1 and si-FOXO1 reversed these effects, promoting cell apoptosis and inhibiting migration (Fig. 7g, h).
Fig. 7
TP53INP1 and P21 act as antitumor effectors mediated by FOXO1 in ESCC cell lines. (a) KYSE150 and TE1 cell lines were transiently transfected with siRNA-TP53INP1, and the cell lysates were subjected to western blotting and Q-PCR. The cell viability was also examined by CCK8 assay. (b) Transwell assay of KYSE150 and TE1 cells transfected with siRNA specific to TP53INP1 (*p < 0.05, ***p < 0.001, ****p < 0.0001.). (c) The expression of TP53INP1 was upregulated by transfecting pCMV-TP53INP1 in KYSE150 and TE1 cell lines. The cell lysates were subjected to immunoblotting and Q-PCR. (d) The apoptosis analysis of TP53INP1 overexpression was examined in ESCC cell lines. (e, f) In the rescue experiment, the siRNA-FOXO1 was transfected singly or co-transfected with pCMV-TP53INP1 into the KYSE150 and TE1 cells. The cell lysates were subjected to western blotting and Q-PCR. (g, h) The rescue experiment under the same transfection conditions analyzed the apoptosis analysis and transwell assay from three independent tests (*p < 0.05, **p < 0.01.). (i) P21 was knocked down by specific siRNA in KYSE150 and TE1 cells, and the cell lysates were subjected to western blotting and Q-PCR. The cell viability was assessed by CCK8 assay. (j, k) A rescue experiment was shown, and the siRNA-FOXO1 was transfected singly or co-transfected with pEnCMV-P21. The cells were cultured for 48 h; then, cell lysates were subjected to immunoblotting and Q-PCR analysis. (l) In the rescue experiment under the same condition, the cells were processed according to the manufacturer’s instructions. The cell cycle profile was obtained from one of three independent experiments. The Flowjo analysis software analyzed the data (*p < 0.05, **p < 0.01.)
P21, as a mediator of cellular proliferation inhibition in response to DNA damage regulated by p53/TP53. In this study, we examined the effect of specific siRNA on P21 in KYSE150 and TE1 cells using western blotting and quantitative PCR. CCK8 assay showed the function of P21 on cell proliferation (Fig. 7i). Subsequently, a rescue experiment was conducted, where overexpression of P21 using pEnCMV-P21 restored the cell cycle progression that was previously induced by FOXO1 deletion (Fig. 7j–l). These findings suggest that P21 overexpression could impede the cell cycle and suppress proliferation in ESCC cell lines.
This study proves that STK3 kinase is an anti-tumor protein in both in vivo and in vitro ESCC. Upregulation of p-STK3/4 was observed to cause the phosphorylation of FOXO1 at Serine212 and the accumulation of FOXO1 in the nucleus, leading to the transcription of downstream genes TP53INP1 and P21 (Fig. 8a).
Fig. 8
STK3 activated FOXO1-TP53INP1/P21 axis in ESCC cell lines and induced cell death. (a) Our study proposes a model of STK3 autophosphorylation in response to oxidative stress. The resulting p-STK3/4 complex regulates FOXO1 phosphorylation at the Serine 212 site and triggers its nuclear accumulation, leading to the transcriptional upregulation of TP53INP1 and P21. This pathway activation induces cell apoptosis and cell cycle arrest in ESCC
留言 (0)