Mouse strain, breeding, and husbandry
Mice were bred and experiments were performed in accordance with the Association for Research for Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Research. Individual study protocols were approved by Stockholm’s Committee for Ethical Animal Research (10389–2018). Animals were housed and fed in a 12 h light/12 h dark cycle with food and water available ad libitum. C57BL/6 J and MitoV [10] mouse strains were bred and utilized at 8–10 weeks of age. For animal group that were treated with NAM, NAM was dissolved in drinking water to achieve a dose of ~ 500 mg/kg/d (based on average water consumption). Water was protected from light and changed every 3–5 days.
Intravitreal rotenone model
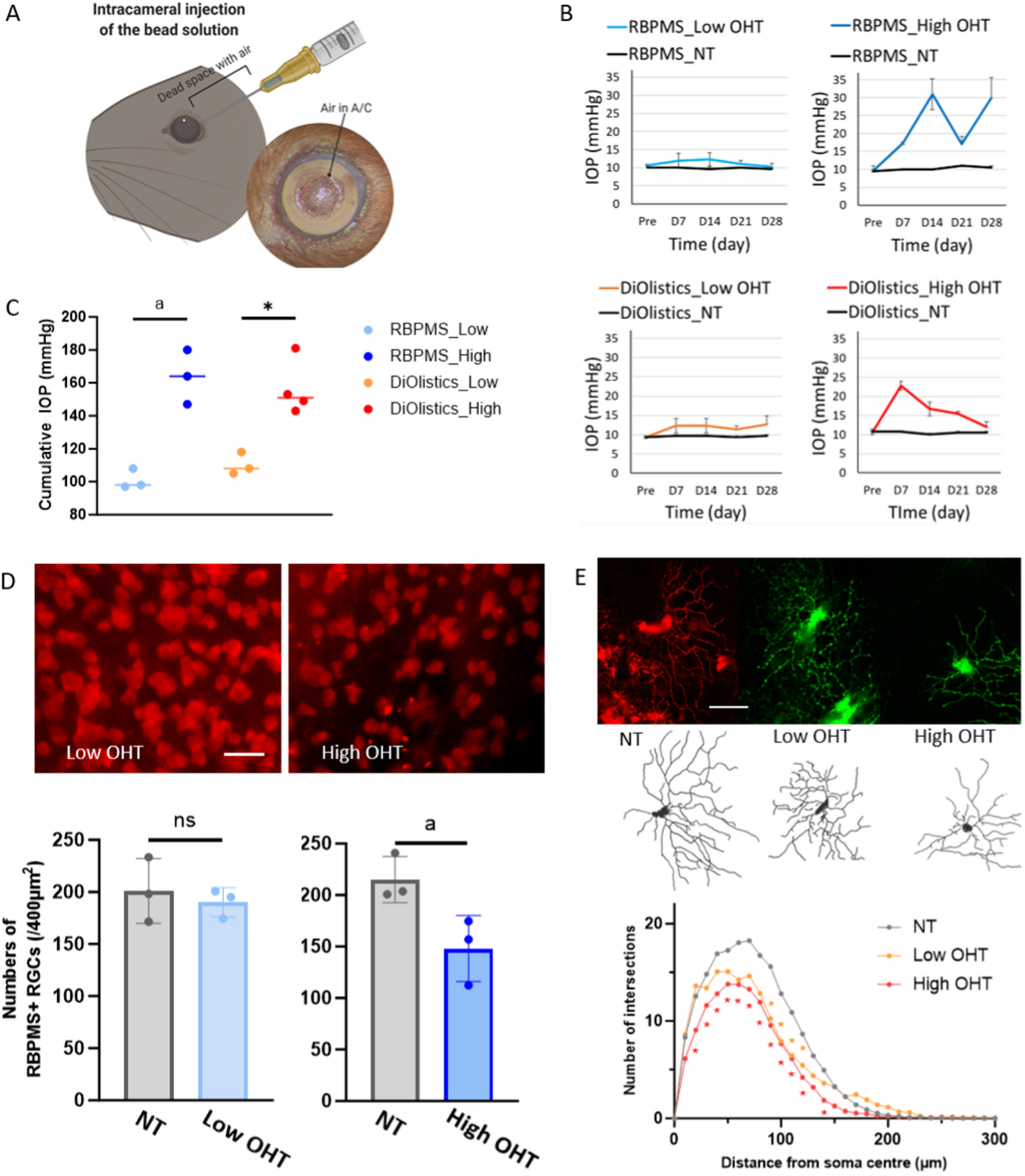
To model a loss of Complex I activity, as in LHON, we used intravitreal injection of rotenone. This model has previously been described [10, 19, 20]. Animals were anesthetized using an intraperitoneal injection of Ketamine (37.5 mg/kg) and Midazolam (Dormitol, 1.25 mg/kg). Bilateral intravitreal injections were performed using a NanoFil 10 μl glass syringe with a 33G needle (WPI). Mice were injected with either 1.5 μL of 10 mM rotenone (MP Biochemicals) in DMSO (Sigma Life science) or 1.5 μL of DMSO only (control). The NAM treated groups underwent one-week of pre-treatment with NAM prior to injection and continuing until mice were euthanized. Mice were euthanized 24 h after intravitreal injection by cervical dislocation and tissues were collected for analysis.
Intravitreal virus injection
Mice were anesthetized as above and intravitreal injection performed using the same equipment. Mice were bilaterally injected intravitreally with 1.5 μL of AAV2-CMV-mCherry (AAV-mCherry; Vectorbiolabs) at 2.7 × 1011 genome copies/mL. Mice were injected with AAV2-mCherry 3 weeks prior to rotenone/DMSO injection.
Cryo-sectioning
Optic nerves were fixed for 24 h in a 3.7% PFA in PBS before immersion in 30% sucrose solution for 24 h. Optic nerves were then embedded in O.C.T. (Sakura) and frozen using dry ice before sectioning (20 μm thickness) in the longitudinal axis using a cryostat (Cryostar NX70, Thermo Scientific). Sections were collected on Superfrost Plus slides (Thermo Scientific) and stored at − 20 °C.
Immunofluorescent labelling
Cryosections and flat mounted retina were subject to immunofluorescent labelling. Cryosections were air dried for 5 min and rehydrated in 1 M PBS for 5 min before following the protocol. Wells were drawn with a hydrophobic barrier pen (VWR), and tissue was permeabilized with 0.5% Triton X-100 (VWR) in 1 M PBS for 20 min, blocked in 1% bovine serum albumin (Fisher Scientific) in 1 M PBS for 45 min, and primary antibody applied and maintained overnight at 4 °C. Primary antibodies used were: anti-RBPMS (Novus, NBP2-20,112, Rabbit, used at 2.2 µg/mL; an RGC specific marker in the retina used only for flat-mounts), anti-GFP (Abcam, ab13970, Chick, used at 20 µg/mL; targeting YFP in MitoV mice), anti-mCherry (Abcam, ab232341, Rabbit, used at 2.02 µg/mL; targeting mCherry in AAV injected mice, used only in optic nerve sections). After 8 washes (15 min each) with 1 M PBS the secondary antibodies were added and incubated at room temperature for 3 h. To limit non-specific binding, 500 µl aliquots of secondary antibodies (at 200 µg/mL) were pre-incubated with a whole PFA-fixed mouse retina for > 48 h. From these aliquots, secondary antibody solutions were prepared at 4 µg/mL for immunofluorescent labelling. Secondary Antibodies used were AF488 (Invitrogen, A11039, goat anti-chick) and AF568 (Invitrogen, A11011, goat anti-rabbit). Secondary only slides were also prepared for all tissues analyzed where only PBS was used (i.e. no primary antibody). Tissues were washed (7 × 15 min) as before and DAPI nuclear stain (1 μg/ml in 1 M PBS) was applied for 10 min. Following a last wash in PBS, glass coverslips were mounted using Fluoromount-G (Invitrogen). Slides and coverslips were sealed with nail-varnish.
Quantification of RGC death
RGC death was quantified in flat-mount retina from MitoV mice (n = 6 eyes for all conditions: DMSO control, DMSO NAM, rotenone control, and rotenone NAM). Images were acquired on an epifluorescent microscope (Leica DMi8). Images were acquired at 40X magnification, (0.25 μm/pixel) and were taken at ~ 1000 μm eccentricity from the optic nerve head. Image locations were at 0, 2, 4, 6, 8, and 10 o’clock about the optic nerve head. Images were cropped to 0.01 mm2 for cell counting. RBPMS + cells and round DAPI nuclei (i.e., not vascular epithelium) were counted using the cell counter plugin for FIJI [21]. For all metrics an average of the 6 crops was calculated for each retina.
Super-resolution fluorescent imaging of mitochondria and morphological analysis
Super-resolution fluorescent images of mitochondria were captured using Airyscan2 imaging on a Zeiss LSM980-Airy (40X/1.2 W, 1,7 X optical zoom, 123.59 × 123.59 μm images, 49 nm/pixel, z-stacks with 19 nm slice thickness). As previously [10], we used Alexa Fluor 488 conjugated secondary antibodies targeting YFP in order to limit signal bleaching of YFP when acquiring image volumes. In flatmount retinas, only the Alexa Fluor 488 signal was captured by Airyscan imaging and RBPMS and DAPI were captured as single slice snaps for reference. In optic nerves, both Alexa Fluor 488 signal (YFP mitochondria) and Alexa Fluor 568 signal (mCherry axons) were acquired by Airyscan imaging. Secondary antibody only controls were used for all experiments to set suitable imaging parameters. For flatmount retinas (n = 6 for all conditions), 2 images were acquired at 1000 μm eccentricity from the optic nerve head. Images were captured as z-stacks from above the NFL to the lower boundary of the IPL (thus capturing all RGC relevant layers). Ganglion cell layer (GCL)/retinal nerve fiber layer (NFL) and IPL were digitally separated as crops for separate analysis. In the retina and optic nerve, mitochondria were reconstructed in 3D using Imaris software (version 9.3.1). Volume reconstructions were performed using the surfaces tool. We used a surface detail of 0.05 µm and a background subtraction of 3 µm for both NFL/GCL and IPL analysis. Signal intensity was subject to a threshold to remove pixels < 800 for the GCL and < 80 for the IPL, followed by an area filter of 0.1. To exclude noise, volumes < 125 voxels were filtered and removed from subsequent analysis. Data for the total number of mitochondria, and the volume, surface area, and sphericity of individual mitochondria were exported. The average value for each of these metrics was calculated per retina. In optic nerves, mCherry filled axons were reconstructed using the surfaces tool and this was used as a mask to exclude mitochondria outside of this axon volume. This allowed the analysis of mitochondria within individual axon bundles.
Electron microscopy
Whole eyes and optic nerves were fixed in 2.5% glutaraldehyde and 1% formaldehyde in 0.1 M phosphate buffer, pH 7.4 at room temperature for 1 h and then stored at + 4 °C. After fixation the samples were rinsed in 0.1 M phosphate buffer pH 7.4 prior to post-fixation in 2% osmium tetroxide in 0.1 M phosphate buffer, pH 7.4 at 4 °C for 2 h. The samples were then stepwise dehydrated in ethanol, followed by acetone and finally resin embedded in LX-112 (Ladd Research). Ultrathin sections (~ 80 nm) were prepared using an EM UC7 microtome (Leica). Whole eyes were sectioned in the sagittal plane to obtain sections through the central retina and optic nerve head (the central optic nerve vessel was used as an anatomic landmark). Cross-sections of optic nerve were collected from the end proximal to the eye. Sections were contrasted with uranyl acetate followed by Reynolds lead citrate. TEM imaging was performed in a Tecnai 12 Spirit BioTwin transmission electron microscope (FEI Company) operated at 100 kV and digital images were acquired using a 2kx2k Veleta CCD camera (Olympus Soft Imaging Solutions). Images of the retina (5 to 25 µm distance from the ONH) were acquired in a series from the inner limiting membrane (ILM) to the lower border of the IPL at 26,500 × magnification. Images of optic nerve were acquired with the same magnification as the retinal images, along two lines across the optic nerve separated by a 90-degree angle.
Ultra-structural measurement of mitochondria
EM images were processed using FIJI. Mitochondria in the NFL/GCL, IPL, and optic nerve were analyzed. For each mitochondrion, multiple morphological metrics were measured: the ratio of the long and short axis length, outer perimeter length, total surface area, cristae number, cristae surface area, the ratio of the cristae surface area to the total surface area, and the mitochondria integrity index. The long and short axis length were measured using the line section tool. The outer perimeter length, total surface area and cristae surface area were measured using the freehand selection tool. The mitochondria integrity index is an index manually measured by counting the number of crossings between the long axis and the cristae [22]. Average values per retina/optic nerve were calculated for all metrics. In the NFL/GCL the number of individual mitochondria were analyzed by condition were: n = 6 DMSO control, n = 6 DMSO NAM, n = 3 rotenone control, n = 6 rotenone NAM. In the IPL the number of individual mitochondria were analyzed by condition were: n = 6 DMSO control, n = 6 DMSO NAM, n = 3 rotenone control, n = 6 rotenone NAM. In the optic nerve the number of individual mitochondria were analyzed by condition were: n = 6 DMSO control, n = 6 DMSO NAM, n = 6 rotenone control, n = 6 rotenone NAM.
Statistical analysis
The statistical analyses were performed in R. Data were tested for normality with a Shapiro Wilk test. Normally distributed data were compared by One-way ANOVA (with Tukey’s HSD). Non-normally distributed were compared by a Kruskal–Wallis test followed Dunn’s tests with Benjamini and Hochberg correction. When comparing individual mitochondrial morphologies (fluorescent or EM) a linear mixed effects model approach [23, 24] was used to account for the higher-level grouping of data (i.e. multiple mitochondria from a single retina) which limits P value inflation whilst limiting the dilution of the heterogeneity of mitochondria within a sample. Conditions were compared individually using this model (lme4 package [25]) and P values were obtained for regression coefficients using the car package. Unless otherwise stated, * = P < 0.05, ** = P < 0.01, ***P < 0.001, NS = non-significant (P > 0.05). For box plots, the center hinge represents the median with upper and lower hinges representing the first and third quartiles; whiskers represent 1.5 times the interquartile range.






留言 (0)