
記住我




The human genome encodes more than 15,000 lncRNAs, which are a highly diverse group of genes. While some lncRNAs encode their function in their sequence, others act as DNA regulatory elements, such as enhancers [55]. Furthermore, it remains unclear how many lncRNAs are functional compared to those that are expressed as transcriptional noise. To better understand the biology of lncRNAs, it is necessary to classify them according to their function.
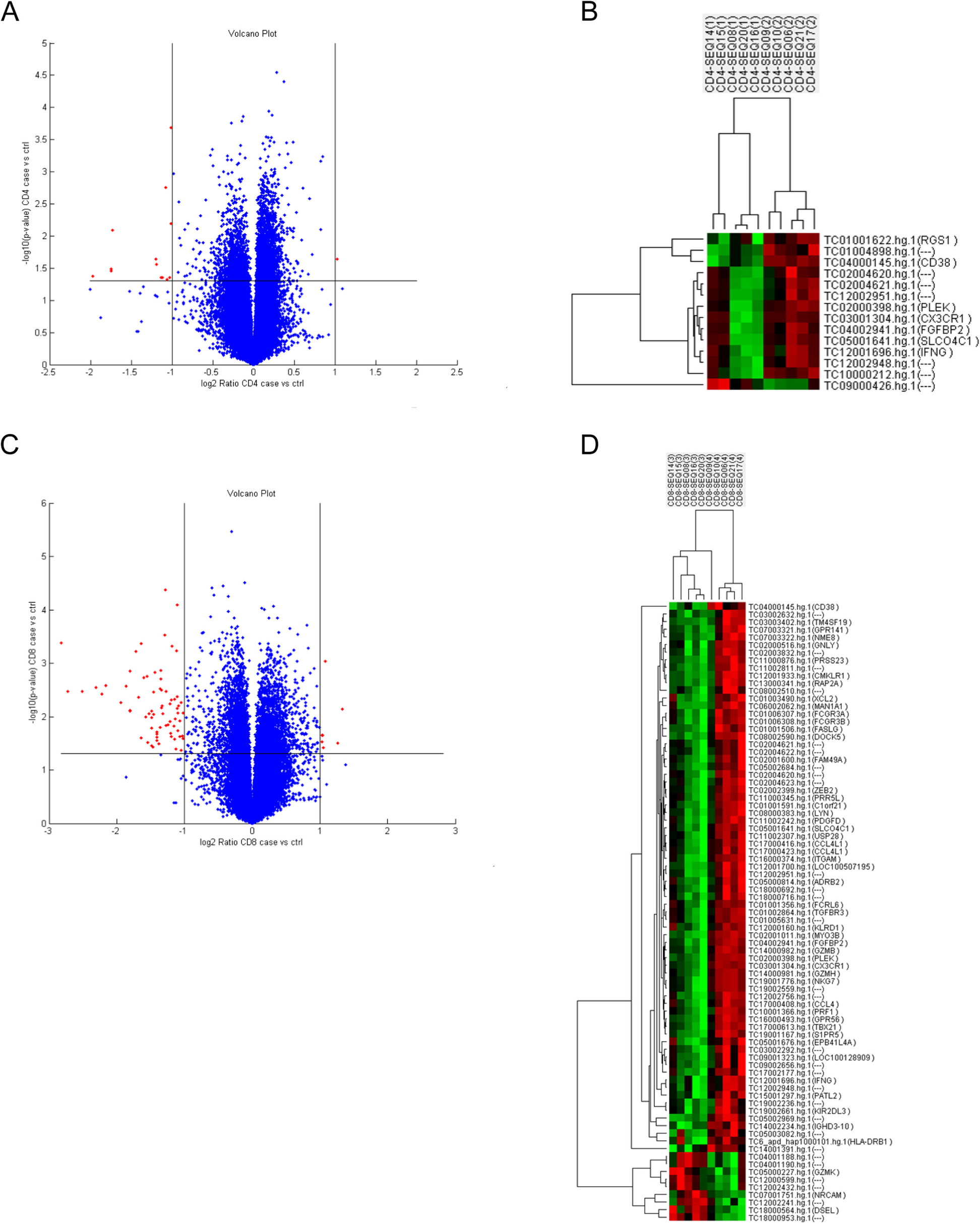
Over the last several years, ML has been exploited to identify complex relationships between biological properties and predict functionality in various contexts [56]. Herein we developed a binary classification model to identify new functional lncRNAs based upon genetic features, which were trained using known functional lncRNAs (Fig. 1a, upper panel).
Fig. 1
Building a machine learning algorithm for the accurate prediction of novel functional lncRNAs. a Workflow for designing the INFLAMeR (Identifying Novel Functional LncRNAs with Advanced Machine learning Resources) algorithm and selecting targets. Upper: The algorithm was trained on previous high-throughput pooled CRISPR interference (CRISPRi) screening data from three previous high-throughput CRISPRi screens [24,25,26]; the algorithm was computed based on a total of 71 features comprising ENCODE ChIP-Seq transcription factor (TF) binding data for the regions surrounding lncRNA promoters [49,50,51] and genomic features. Lower: targets predicted to be functional by INFLAMeR in the K562 cell line (INFLAMeR score > 0.5) were selected for validation after excluding lncRNAs that were functionally characterized in previous studies, those with low expression, those not annotated in the Ensembl database, and those neighboring a protein-coding (PC) gene; thirty-nine lncRNAs were selected for validation. b INFLAMeR was built using an XGBoost classifier and its performance was calculated using a receiver operating characteristic (ROC) curve. Black ROC curve shows the mean classifier performance on the test set using three randomized seeds (red, green, and yellow curves). c Confusion matrix based on the test set. Percentages in the confusion matrix are row-normalized. d Local explanation summary of the impact of the top twenty features on the INFLAMeR score. Each dot represents one lncRNA. Red indicates a higher feature value (e.g., larger transcription start site (TSS) protein-coding (PC) distance or higher expression), and blue indicates a lower feature value (e.g., smaller TSS PC distance or lower expression). Higher (red) feature values with a positive SHAP value indicate a positive correlation, and lower (blue) feature values with a positive SHAP value indicate a negative correlation
To create the ML algorithm, we gathered 143 genetic variables representing different features of lncRNAs (Additional file 1: Table S5). These features included eighteen categorical variables related to lncRNAs and their surrounding genetic regions, such as the transcript length, number of exons, proximity to enhancers, and proximity to PCGs. While these features are important, most do not take into account the cell type-specific functions of lncRNAs. Since most lncRNAs exhibit cell type-specific functionality [1, 5], we also included features pertaining to regulation and expression, namely TF binding data from the ENCODE project for lncRNA promoters in four different cell lines for more than 100 TFs [49,50,51]. We then trained the ML algorithm using a set of functionally validated lncRNAs. Although many human lncRNAs have been demonstrated to be functional in various contexts, such as cell proliferation [24, 57], differentiation [26, 58], and disease [20, 59, 60], these lncRNAs have often been studied in different cell types and under varying experimental conditions, resulting in scattered data.
In recent years, high throughput reverse genetic screens have been increasingly utilized to explore the functions of numerous lncRNAs [61]. These screens, which involve pooled perturbations, offer a major advantage as they allow thousands of genes to be tested in parallel under identical experimental conditions. The resulting scores generated by these screens are statistically based and enable the classification of genes as either hits (functional) or non-hits. While various perturbation methods have been employed to identify functional lncRNAs, CRISPRi has emerged as the most applied technique across multiple cell types and biological contexts [1].
To train our algorithm, we incorporated data from three genetic screens [24,25,26] that targeted 16,401 lncRNAs across seven cell lines. However, these data are imbalanced since only 9% (n = 1451) of the lncRNAs were identified as functional. Moreover, perturbation screening of lncRNAs is subject to high false-negative rates due to the inconsistent efficiency of CRISPRi [62] and poor annotation of lncRNA TSSs. This dataset imbalance may introduce a bias into the training algorithm, resulting in diminished predictive value.
To develop the ML model, we experimented with three different cost-sensitive classifiers: logistic regression, balanced random forest, and extreme gradient boosting (XGBoost). We assessed the performance of the model using a range of metrics, including AUROC, sensitivity, specificity, F1-score, precision, and Brier score (Additional file 1: Table S6). Our results show that the mean AUROC for logistic regression was 0.778, which was lower than that for both XGBoost and balanced random forest (mean AUROC values: 0.8236 and 0.8335, respectively; Additional file 1: Fig. S1c).
To determine whether XGBoost or balanced random forest would be better suited for our desired balance of sensitivity and specificity, we determined the percentages of true positive and true negative results for each method (Additional file 1: Fig. S1d). XGBoost had a somewhat higher specificity percentage than balanced random forest (82.24% vs. 80.84%). On average, both XGBoost and balanced random forest produced a similar number of true positive cases (66 and 69 cases, respectively). Additionally, XGBoost possessed two important advantages over balanced random forest. Firstly, in terms of metrics, XGBoost had higher F1-score and precision outcomes (0.1264 and 0.0693, respectively) than balanced random forest (0.1240 and 0.0675, respectively). Secondly, XGBoost had a shorter training time than balanced random forest, which enabled us to experiment and explore more training settings. Therefore, we selected XGBoost for our ML model.
To compensate for the class imbalance problem in our dataset, we tried two different strategies. Initially, we applied random under-sampling of non-hits with and without replacement as preprocessing before training the XGBoost model. We experimented with different sampling strategies to find the optimal ratio of non-hits to hits. The sampling strategies were as follows: 3%, 4%, 5%, 10%, 20%, 30%, 40%, and 50% without and with replacement (Additional file 1: Fig. S1e and f, respectively). We evaluated the performance of each strategy using various metrics such as accuracy, precision, recall, F1-score, and AUROC. We obtained the best performance using the 50% sampling strategy (1822 non-hits and 911 hits), both without and with replacement (Additional file 1: Tables S7 and S8, respectively). We next explored a cost-sensitive approach that assigns different weights to the classes based on their proportion within the dataset, where the algorithm was adjusted to favor the detection of the minority class. This required a modification of the optimization function in the training step of the learning algorithm. For this approach, we implemented several different modifications of the scale_position_weight and class_weight values. First, we ran the algorithm without a cost value; this means the majority and minority class had the same weight. Second, we applied the default scale position weight, defined as sum(majority class)/sum(minority class). Finally, we applied a scale_position_weight/class_weight of 100 and 1000. A value of 100 showed the best result, and was used to compare the cost-sensitive XGBoost model with the under-sampled XGBoost models. We found that the cost-sensitive model exhibited superior performance compared to under-sampling with 50% in terms of the AUROC and sensitivity values. Thus, cost-sensitive XGBoost was selected as our ML model.
We used Shapley (SHAP) values [29] to assess the importance of each feature in our XGBoost model for predicting lncRNA function. SHAP values are a useful metric for understanding how a ML model learns from the input features from a global and individualized perspective. SHAP values have several distinct advantages over other explainability approaches such as the gain and split decision tree methods. Firstly, the SHAP approach is model agnostic, meaning the same framework can be used for many different models in addition to XGBoost. More importantly, SHAP values explain the output of a function f(x) as a sum of the effects (φ) of each feature being introduced into a conditional expectation. Additionally, for nonlinear functions, the order in which features are introduced affects the output. SHAP values are given as the average of all possible orderings, whereas the gain and split methods are inconsistent because they only consider a single ordering [63]. Out of the 143 features, 30 had no predictive value (SHAP ≈ 0) and were hence excluded from further analysis. Although combining multiple features can enhance the predictive power of the model, it can also introduce high-dimensional redundancy and increase the training time and performance bias [64, 65]. Therefore, we applied an RFE method based on SHAP values to select the optimal subset of features that balanced sensitivity and specificity on the test set. Based on RFE, the best performance was obtained with 71 features (Additional file 1: Fig. S1g).
We then evaluated our cost-sensitive XGBoost model with RFE using a triple-repeated tenfold cross-validation with stratified sampling. The hyper-parameters of our gradient-boosted tree classifier were as follows: initial guess of 0.5, gamma of 1.0, gain as importance, learning rate of 0.05, residual-trees with 5 depth levels and 28 leaves (Additional file 1: Fig. S1b), 100 residual-trees, random seed of 0, regularization of lambda of 5.0, and a cost ratio of 100 for misclassifying a hit versus a non-hit.
Using 71 features, the cost-sensitive XGBoost method showed better performance than previous methods. In fact, all metrics were higher for the 71-feature model compared to the 143-feature model; sensitivity and AUROC were increased by 0.05 and 0.002, respectively (Additional file 1: Table S9). We also compared the final XGBoost model to our previous approach using balanced random forest. The XGBoost model with RFE displayed higher mean values of specificity, F1-score, and precision (0.8227 vs. 0.8084, 0.1275 vs. 0.1240, 0.0698 vs. 0.0675, respectively) than the balanced random forest model (Additional file 1: Table S9). Although it did not exhibit the highest score in all metrics, the most important metrics for our analysis—namely the true positive and true negative values, and their balance—were the highest using this strategy, while the rest of the metrics were supplemental.
To estimate the probability of hits among all lncRNAs, we applied our trained model to classify the 50,847 transcripts in our dataset. The INFLAMeR score was given as a value between zero and one, defined as the probability of the transcript to be functional. Transcripts with a score greater than 0.5 were considered to be predicted hits for our analysis. To evaluate the performance of our model, we used a ROC curve and a confusion matrix. Our model achieved a mean AUROC of 0.8250 ± 0.01 (with minimum and maximum values of 0.78 and 0.87, respectively, Fig. 1b; see Additional file 1: Table S10 for all AUROC values), with a true positive rate of 72.92% and a true negative rate of 82.27% (Fig. 1c). Additionally, lncRNAs previously identified as hits tended to have a higher INFLAMeR score (Additional file 6: Table S11), consistent with the high true positive rate.
Statistical analysis of the impact of individual features on the score confirmed the importance of lncRNA-PCG distance (Fig. 1d). Interestingly, the impact of the distance on the prediction score was not linear; rather, we observed a decrease in the score in a stepwise manner, with the SHAP values sharply decreasing around 1000 and 2000 bases from the nearest PCG. However, the distance between promoters did not further affect the score beyond 2000 bases (Additional file 1: Fig. S1h). Further important features were those affecting transcription, such as transcription level (defined as log FPKM), the number of TFs binding to the promoter, and the binding of specific TFs including SIN3A (Fig. 1d). For example, the score of the lncRNA SNHG6 was decreased owing to its distance from a PCG and proximity to a traditional enhancer; however, it was increased based on its high expression and SIN3A binding at its promoter (Additional file 1: Fig. S1i).
Interestingly, although transcription was tightly correlated with lncRNA functionality (Wilcoxon test, p = 4.20 × 10–222), the presence of general TFs such as TAF1 and TAF7 did not contribute to the prediction model. These findings suggest that functional lncRNAs may be regulated by a subset of specialized factors.
Our findings indicate that lncRNAs have distinctive features that are closely linked to their functionality. These attributes can be influenced by various factors, such as their genomic location (for example, their proximity to PCGs) and by cell type-specific mechanisms that are intricately regulated by TFs. By identifying these characteristics, one can gain a deeper understanding of the complex roles that lncRNAs may play in regulating genes and cellular processes.
Validation of INFLAMeR’s predictions and identification of functional lncRNAsTo assess the accuracy of INFLAMeR, we selected a subset of lncRNAs for CRISPRi-mediated knockdown (KD) in K562 cells based on their INFLAMeR score and according to several cutoff parameters (Fig. 1a, lower panel). We excluded the following lncRNAs for validation: those identified in previous screens to be functional in K562 cells; those with low expression (Log2 FPKM < 0) in K562 cells; those which are not annotated in the Ensembl database; and those located within 1 kb of the nearest PCG TSS, as transcription can be initiated from divergent promoters. We included thirty-nine lncRNAs predicted to be functional in K562 cells, with an INFLAMeR score ≥ 0.5. Furthermore, we randomly selected seven lncRNAs that did not meet the INFLAMeR threshold for functional prediction (INFLAMeR score < 0.4) to validate their non-functionality using the same cutoff parameters mentioned above. This was done in order to exclude the possibility that the selected lncRNAs were functional by chance.
Predicting CRISPRi-mediated KD efficiency can be challenging, especially for lncRNAs with multiple splice variants. To enhance KD efficiency, we pooled sgRNAs for each target, with each sample containing two sgRNAs targeting the same lncRNA [66]. We confirmed efficient KD, defined here as relative expression ≤ 0.5 by qPCR, for all the targeted lncRNAs (Additional file 1: Fig. S3a).
To determine whether the validated lncRNAs affected cell proliferation upon KD, we conducted a two-color CCG assay for each sample (Fig. 2a); briefly, cells with KD of each lncRNA (or transduced with a non-targeting sgRNA) expressing BFP were mixed at a 1:1 ratio with cells transduced with a non-targeting sgRNA expressing GFP, and the relative proportion of BFP-positive cells was measured over 14 days by flow cytometry to determine the impact of KD on cell proliferation. We observed a significant reduction in the proportion of BFP-positive cells (p < 0.05) for 74% of the samples predicted to be functional (n = 29/39), while none of the seven lncRNAs predicted to be non-functional had a significant impact on cell proliferation (Fig. 2b).
Fig. 2
Most lncRNAs predicted to be functional showed a functional effect upon knockdown (KD). a Two-color competitive cell growth (CCG) assay. Left: K562 cells stably transduced with sgRNAs targeting a given lncRNA (or non-targeting sgRNA control)—including a blue fluorescent protein (BFP) marker gene—were mixed at a 1:1 ratio with cells transduced with sgRNA control—including a green fluorescent protein (GFP) marker gene—and the fraction of BFP-expressing cells was tracked over 14 days by flow cytometry. The effect of KD on cell proliferation was expressed as the fraction of BFP-expressing cells relative to that at day 0. Middle: a representative example of the change in the relative fraction of BFP-expressing cells throughout the experiment, normalized to day 0. Right: Representative examples of the BFP- and GFP-expressing fractions based on flow cytometry throughout the experiment. b The relative growth of BFP-expressing cells after transduction with the sgRNA control (black), thirty-nine lncRNAs predicted to be functional by INFLAMeR (green), and seven lncRNAs predicted to be non-functional (blue) at day 14 of the CCG assay. Error bars represent SD (n = 3 biological replicates). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. sgRNA control using a one-tailed t-test. c Relative cell survival after 72 h incubation with 1 µM daunorubicin (DNR)—relative to that in untreated cells—for sgRNA control (black), thirty-nine lncRNAs predicted to be functional (purple), and seven lncRNAs predicted to be non-functional (blue). Error bars represent SD (n = 3 biological replicates). *p < 0.05, **p < 0.01, ***p < 0.001 vs. sgRNA control with Bonferroni correction. (d) Of the thirty-nine predicted lncRNAs, many of those with an effect on cell proliferation (green) also affected resistance to DNR (purple) upon KD
Importantly, we did not observe any enhancement in cell proliferation following the knockdown of lncRNAs. This outcome aligns with the intrinsic characteristics of our cell model, derived from leukemia, where cells are already optimized for maximal proliferation under favorable growth conditions. Hence, the knockdown of lncRNAs is more likely to manifest as a reduction rather than an enhancement of proliferation rates.
Since lncRNAs often function in cis, we investigated whether the validated genes were adjacent to PCGs considered essential based on dependency scores obtained using the DepMap portal, which characterizes essential genes based on gene perturbation across more than 1000 cancer cell lines [67]. We found no correlation between the INFLAMeR score of the thirty-nine predicted lncRNAs and the essentiality of their neighboring PCGs, further supporting the independent function of the lncRNAs (Additional file 1: Fig. S4).
The observed effect on cell fitness may be the cumulative result of alterations in cell cycle and cell viability. Therefore, we further analyzed changes in cell cycle and apoptosis for eleven lncRNAs that showed the most dramatic effect on cell proliferation. We observed significant changes in the cell cycle for nine lncRNAs (Additional file 1: Fig. S5a). In agreement with this finding, we also observed an increase in the fraction of apoptotic cells in six of these samples (Additional file 1: Fig. S5b), as well as increased basal DNA damage in nine samples, as determined based on increased levels of the bona fide DNA damage marker γH2AX (Additional file 1: Fig. S5c).
However, CRISPRi may result in off-target effects owing to the genomic positioning of the target gene or low-specificity interactions of the sgRNA caused by sequence similarity. Therefore, to validate the on-target specificity of our approach, we performed TSS deletion using Cas9 with dual sgRNAs. To this end, we tested three pairs of sgRNAs flanking the TSS for each of the top ten lncRNAs. For most genes (n = 8/10), we achieved a significant reduction in lncRNA expression from one sgRNA pair (Additional file 1: Fig. S3b). Importantly, most of these cells showed the same impact on cell cycle regulation (Additional file 1: Fig. S6a) and apoptosis (Additional file 1: Fig. S6b) as those observed after CRISPRi, indicating the gene-specific effect of our findings.
Studying cell proliferation under optimal growth conditions may overlook lncRNAs that function in response to stress cues. Therefore, we incubated each sample with DNR, an anthracycline chemotherapeutic agent commonly used to treat leukemias [34]; DNR exerts its pharmacologic activity as a DNA topoisomerase II inhibitor as well as a DNA intercalator. After confirming the optimal concentration of DNR for the growth inhibition assay based on control cells and ten lncRNAs (Additional file 1: Fig. S7; see methods), each sample was incubated with 1 µM DNR for 72 h to determine whether these lncRNAs affected DNR activity. Of the thirty-nine hits, 44% (n = 17/39) exhibited a significant effect on DNR activity (Fig. 2c), while the seven non-hits did not affect DNR resistance. Of the seventeen lncRNAs affecting DNR resistance, thirteen also showed a significant reduction in cell proliferation from the CCG assay (Fig. 2d).
Thus, 85% (n = 33/39) of the lncRNAs predicted by INFLAMeR to be functional were experimentally validated to have a function in either cell proliferation or anticancer drug resistance. Importantly, most of these lncRNAs have never been functionally characterized until now, and none of them has a known function in K562 cells.
Collectively, these findings demonstrate the utility of INFLAMeR in predicting the functionality of lncRNAs.
Characterization of top lncRNAs affecting cell proliferationTo further elucidate the functional mechanisms of nine lncRNAs significantly influencing cell proliferation upon KD, we performed transcriptome analyses. Principal component analysis (PCA) displayed distinct clustering for each lncRNA (Fig. 3a). Notably, for most lncRNAs, we identified a relatively modest number of highly differentially expressed genes (DEGs), ranging from 70 to 300 DEGs—characterized as having a |Log2 Fold Change|> 0.7 and a probability > 0.75. Additionally, analyzing the most variable DEGs for each lncRNA revealed unique gene expression patterns in the samples (Fig. 3b, Additional file 1: Fig. S8). This suggests that although KD of each lncRNA hindered cell proliferation, it likely occurred through distinct mechanisms. We also noted that for all nine lncRNAs under study, gene expression was impacted across the genome (Additional file 1: Fig. S9).
Fig. 3
Transcriptome analysis for nine validated lncRNAs. a Principal component analysis was performed for the top variable genes in each sample to confirm that each lncRNA affected a distinct group of genes upon knockdown (KD). b Differential gene expression analysis indicated that most of the lncRNAs affected a unique subset of genes upon KD
LncRNAs can affect local transcription (cis) or operate via distal interactions (trans). To gain a deeper understanding of their functions, we overexpressed ten of these lncRNAs through pseudo-lentiviral transduction (Additional file 1: Fig. S3c). Overexpression of these lncRNAs led to a partial or complete rescue of cell cycle regulation and apoptosis (Additional file 1: Fig. S10a and b, respectively). Among the genes investigated, SNHG6 emerged as an especially noteworthy candidate.
SNHG6 regulates hematopoiesis in K562 cellsSNHG6 is a lncRNA with a known impact in several cancer types [68,69,70,71,72,73,74], and is significantly associated with a poor prognosis in acute myeloid leukemia (Fig. 4a).
Fig. 4
Small nucleolar host gene 6 (SNHG6) acts as a regulator of hematopoiesis. a High SNHG6 expression is associated with reduced overall survival in acute myeloid leukemia. b SNHG6 knockdown (KD) affects the expression of genes across the genome, implying a trans regulatory function. Blue represents downregulation, red represents upregulation. c SNHG6 KD led to the upregulation of the platelet-associated genes PPBP and PF4, and the erythrocyte-associated genes AHSP and LYAR. d Transcription factors (TFs) strongly associated with the differentially expressed genes (DEGs) upon SNHG6 KD include hematopoiesis-associated TFs such as CEBPZ, GATA1, TAL1, and KLF1. e Gene Ontology (GO) analysis of the DEGs indicates a strong correlation with erythrocyte differentiation. f GO analysis of the TFs known to bind SNHG6 in K562 cells shows a strong association with myeloid differentiation- and hematopoiesis-related pathways
The KD of SNHG6 led to the differential expression of 304 genes, including 126 protein-coding genes and 103 lncRNAs. The identified DEGs were widespread across the entire genome (Fig. 4b), suggesting a trans regulatory role for SNHG6. This was consistent with the cytoplasmic localization of SNHG6 in K562 cells (Additional file 1: Fig. S11), and the observed phenotypic rescue caused by overexpression (Fig. 5a and Additional file 1: Fig. S10).
Fig. 5
SNHG6 KD promotes erythrocyte differentiation and prevents megakaryocyte differentiation in K562 cells. a Cell proliferation was significantly reduced by both knockdown (KD) and knockout, while overexpression in KD samples restored proliferation rates. b GPA levels were normalized to those in untreated cells in each sample to quantify the degree of erythrocyte differentiation. SNHG6 KD promoted erythrocyte differentiation. Error bars represent SD (n = 3 biological replicates). *p < 0.05, **p < 0.01 vs. sgRNA control. c CD41/CD61 levels were normalized to basal levels in each sample to quantify the degree of megakaryocyte differentiation. SNHG6 KD significantly reduced megakaryocyte differentiation after 72 h. Error bars represent SD (n = 3 biological replicates). **p < 0.01 vs. sgRNA control. d–f SNHG6 KD led to reduced tumor volume (e) and weight (f) in a xenograft mouse model. d Representative tumors. Error bars represent SEM. *p < 0.05 vs. sgRNA control by one-way ANOVA (n ≥ 11 from two independent experiments). g Proposed model for the mechanism of function of SNHG6
Interestingly, SNHG6 KD led to the upregulation of genes involved in myeloid differentiation to both erythrocyte and megakaryocyte lineages. Following SNHG6 KD, the most strongly up-regulated genes included the erythrocyte-associated α-hemoglobin stimulating protein (AHSP) and Ly1-antibody reactive (LYAR); and the megakaryocyte-associated pro-platelet basic protein (PPBP) and platelet factor 4 (PF4) (Fig. 4c). Using ChEA3, a tool for identifying the TFs most strongly associated with the DEGs [45], we observed a strong correlation with several TFs associated with hematopoietic differentiation, including CEBPZ [75], KLF1, TAL1, and GATA1 [76] (Fig. 4d). Consistent with the erythroleukemia origin of K562 cells, GO analysis using Enrichr [42,43,44] showed strong enrichment for erythrocyte differentiation (Fig. 4e).
We also performed GO analysis of the TFs targeting the SNHG6 promoter. Expectedly, the most strongly enriched pathways were associated with transcription (Additional file 1: Fig. S12). Surprisingly, however, there were several highly enriched pathways relating to hematopoietic and myeloid differentiation (Fig. 4f). These findings are in line with recent reports showing that SNHG6 plays a role in myeloid cell differentiation in mice, and computational predictions suggesting a role in leukemia progression and patient prognosis [70,
留言 (0)