
記住我

A phase 2 open-label study was conducted at the NIH (NCT03571191). This investigator-sponsored study was supported by Amgen, Inc.; the study design, conduct, and analyses were performed by the investigators. The subjects received denosumab for 6 months at a dose of 120 mg every 4 weeks, with loading doses administered on weeks 2 and 3.30 Percutaneous FD lesion biopsies were performed at baseline and at 6 months in 6 of the 8 total adult subjects (Supplementary Fig. 1). Biopsies were deferred in 2 subjects: 1 due to the presence of isolated craniofacial FD and 1 due to intercurrent illness unrelated to the study intervention. Biopsy sites were chosen jointly by the investigators and surgeons to ensure that the procedure was minimally invasive, and biopsies were performed in the interventional radiology suite at the NIH Clinical Center using core needles under CT guidance. The same sites were biopsied before and after denosumab treatment.
In vivo and ex vivo models of fibrous dysplasiaMice expressing the GαsR201C variant were generated as previously described.8 At 10 weeks of age, GαsR201C expression was induced in the limbs by switching to doxycycline-supplemented food (100 × 10−6 Purina Mod LabDiet 5001; PMI Nutrition International, Saint Louis, MO). Six mice received an αRANKL antibody (6 mg/kg, BE0191, Bioxcell Lebanon, NH). Six GαsR201C mice as well as 6 littermate controls not harboring tet-GsαR201C received a rat IgG2A isotype control (6 mg/kg, BE0089, Bioxcell). All mice received subcutaneous injections on Days 28, 30, 35, 42, 49 and 56 after induction. Plasma was collected weekly, and on Day 58, the mice were euthanized, and tissue was collected. FD or control tissue was extracted from the distal ulna and radius, cleaned from the muscle and tendons, and snap-frozen for RNA analysis. Mice were then perfused with PBS and Z-fix fixative, and both hindlimbs were extracted for histology and µCT analysis.
GαsR201C expression was induced in ex vivo bone marrow cultures,11 which were subsequently treated with an αRANKL antibody. MC3T3-E1 clone 4 and 14 cells were cultured as previously described.31
Mouse plasma measurementsMouse blood (120 µL) was collected weekly via retro-orbital eye collection. At the time of euthanasia, 500–1 000 µL was obtained from the vena cava. Blood was stored in heparinized vials, and plasma was obtained. TRAP5b, CT-X and P1NP were measured using IDS ELISA kits SB-TR103, AC-06F1 and AC-33F1, respectively.
Mouse X-rays and microCTMice were anesthetized with 2%–5% isoflurane, and X-ray images of the hind limbs were obtained on a Faxitron Ultrafocus system (Hologic, Marlborough, MA). A semiquantitative score was developed and validated to quantify the disease burden (Table S1). Three examiners independently evaluated the X-rays of both hindlimbs of each mouse at each timepoint.
The right hindlimbs were dissected and scanned using a Scanco µCT 50 at 10 µm, 70 kVp, 80 µA, and 900 ms integration time (Scanco, Wangen-Brüttisellen, Switzerland). The reconstructed images were analyzed with Analyze 14 (AnalyzeDirect, Overland Park KS) and calibrated against hydroxyapatite phantoms of known density. The volume of interest (VOI) was defined as the distal tibia sector between 500 µm below the fibula insertion point and 200 µm above the intermedium. The SmartTrace tool was used to outline the distal tibia every 50 µm in the sagittal direction, and then, the Propagate Objects tool was used to connect the 2D tracings, semiautomatically segmenting the VOI.
Following segmentation, the average bone mineral density (BMD) in milligrams of hydroxyapatite per cubic centimeter (mg HA/cm3) was obtained for the entire VOI. The distal tibiae were further analyzed by obtaining the number of voxels per BMD unit and binned into three categories: soft tissue (<350 mg HA/cm3), partially mineralized tissue (350-600 mg HA/cm3), and mineralized tissue (>600 mg HA/cm3).
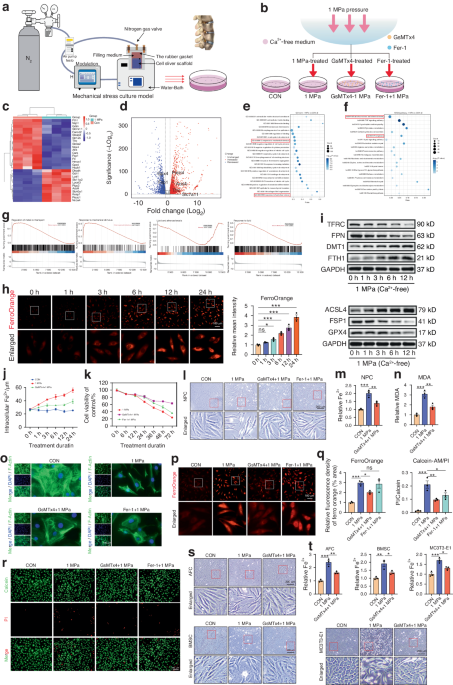
RNA extraction and sequencingMouse and human samples were snap-frozen, pulverized using an automated dry pulverizer (CP02 cryoPREP, Covaris, Woburn MA) and immediately transferred to TRIzol (Thermo Fisher Scientific). Phenol‒chloroform RNA extraction was carried out. cDNA synthesis, library construction and sequencing were performed by Novogene (University of California, Davis, CA). Briefly, messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, first-strand cDNA was synthesized using random hexamer primers, followed by second-strand cDNA synthesis, end repair, A-tailing, adapter ligation, size selection, amplification, and purification. The library was assessed with a Qubit fluorometer and real-time PCR for quantification and a bioanalyzer for size distribution detection. The quantified libraries were pooled and sequenced on an Illumina NovaSeq 6000 sequencer according to the effective library concentration and data amount in 150 ×150 paired-end mode. After sequencing, the base-called demultiplexed (fastq) read qualities were determined using FastQC (v0.11.2)32 and aligned to the GENCODE v32 human genome (GRCh38.v32) from human samples and the GENCODE M23 mouse genome (GRCm38. M23) for mouse samples and gene counts generated using STAR (v2.7.3a).33 Datasets are availble at the NCBI GEO repository with series accession number GSE250357. Postalignment qualities were generated with Picard tools. An expression matrix of the raw gene counts was generated using R34 and filtered to remove genes with low counts (defined as those with fewer than 5 reads in at least one sample). The filtered expression matrix was used to generate a list of differentially expressed genes between the sample groups using DESeq2,35 and the genes were analyzed via principal component analysis (PCA). Reads corresponding to chromosomal regions chr20:909,365 and chr20:58,909,366 were analyzed to detect and quantify the GNAS p.R201C (C > T) and pR201H (G > A) substitutions, respectively. An FD tissue genetic signature comprising 276 genes was derived by comparing WT versus Gnas R201C-expressing mice; the genes had a > 2 log2-fold change and were significantly (adjusted P value ≤ 0.05) differentially expressed between the two groups. This gene signature was converted to human genes based on homology scores using the Biomart service, which reduced the number of genes to 202. Similarly, a list of genes positively correlated with the proliferation marker PCNA in healthy human tissues was compiled from Venet et al.10 A custom.gmt file was compiled from the FD, meta-PCNA signature and MSigDB GO-derived gene sets corresponding to osteoclast and osteoblast differentiation and activity biological processes. For osteoblasts, the gene set GO:0001649 “osteoblast differentiation” list was used. For osteoclasts, we combined two gene sets: GO:0030316 “osteoclast differentiation” and GO:0045453 “bone resorption”. The gene set variation analysis (GSVA)36 method was used to compute enrichment scores for each sample against these selected gene sets using the Poisson kernel, and the resulting matrix was subjected to unsupervised clustering using the Euclidean metric. Selected gene expression heatmaps were generated from log-transformed TMM-normalized gene counts using a heatmap.2R function.
Ex vivo FD culturesBone marrow explants were isolated and cultured as described previously.11 Briefly, the tibiae and femurs were dissected from uninduced mice or wild-type littermates, the marrow cavities were opened by drilling small holes in the epiphyses using hypodermic needles, and the marrow was flushed using a syringe containing culture media. The isolated marrow was then triturated and added to a T-75 culture flask supplemented with complete explant media (α-MEM supplemented with 20% fetal bovine serum and 1x penicillin/streptomycin) supplemented with 1x Normocin (ant-nr-1; InvivoGen) during culture establishment. The cells were left undisturbed in a cell culture incubator for 10 days, after which a mixed culture of predominantly BMSCs and osteoclast precursors was established. Cultures were subsequently passaged via 0.05% trypsin washing, lifted via cell scrapers and used for downstream experiments. Cultures below passage 4 were used for the experiments. For FD induction, cells were plated at ~40% confluency in 6-well plates, 24-well plates, or angiogenesis µ-slides (Cat No. 81506 Ibidi, Firchburg, WI) and treated with 5 µM doxycycline (Sigma, # D9891-5G) to activate GαsR201C-expressing BMSC lineage cells. During induction, the media were refreshed daily. Initial osteoclast fusion was typically observed after 3 days of induction, and samples were collected after 4 days of induction. Neutralizing RANKL antibody (BE0191, Bioxcell) or IgG2a (BE0089, Bioxcell) was added at 1 µg/mL at the beginning of induction, after which the cells were maintained throughout the experiment. MC3T3-E1 clones 4 and 14 were cultured as previously described.31
The human BMSCs cultured for BaseScope optimization were cultured as previously described.15
Histology and cell culture stainingTissue and culture preparationDissected mouse hindlimbs and human baseline and post-treatment bone biopsies were fixed in Z-fix (Anatech, USA) overnight at 4 °C. Samples for paraffin embedding (PE) were decalcified in 0.25 mol/L EDTA at 4 °C. The samples were then embedded and sectioned into 5 µm sections. The slides were stored at 4 °C or –20 °C until staining. Sections from all PE samples were deparaffinized in xylene and rehydrated using a graded ethanol series for subsequent staining. Tissue morphology was assessed by staining sections with H&E.
Tissue samples for cryosectioning were shipped to Bonebase (at UConn Health, Farmington, CT) in formalin and in wet ice and embedded and cryosectioned following the methods of Dyment et al.37
The cell cultures were fixed with warm, freshly prepared 4% formaldehyde in PBS (Sigma, F1268) at 37 °C.
Patient and healthy volunteer-derived BMSC cultures were pelleted at 4 °C and 300 × g for 5 min, embedded in OCT media, frozen and cryosectioned for Gαs mRNA detection, optimization and validation.
TRAP and ALP enzymatic detectionMouse cryosections were stained for TRAP and ALP enzymatic activity at Bonebase following previously described methods.37 Mouse culture TRAP staining was performed with a Cosmo Bio TRAP Staining Kit (Cat No. PMC-AK04F-COS, Cosmo, Carlsbad, CA). Human PE sections were incubated with TRAP staining solution (Wako, Cat No. 294-67001) for 25 min at RT, counterstained with Methyl Green and mounted using EcoMount.
ImmunohistochemistryPE sections were deparaffinized in xylene and rehydrated using a graded ethanol series. Endogenous peroxidase activity was blocked using 3% H2O2 in methanol. For Runx2 and MCM2, antigen retrieval was performed using a Uni-Trieve (Innovex Biosciences, Cat No. NB325) for 30 min at 55 °C or 45 min at 70 °C, respectively. Nonspecific binding was blocked using goat or rabbit serum (Vector, Cat No. PK-6105 and PK-6101) as appropriate. Next, both human and mouse sections were incubated with rabbit anti-Runx2 (1:400; Abcam, Cat No. ab192259), or rabbit anti-Mcm2 (1:200; Abcam, Cat No. 108935); human sections were incubated with rabbit anti-osteocalcin (1:200; Proteintech, Cat No. 23418-1-AP); and mouse sections were incubated with goat anti-sclerostin (1:100; R&D Systems, AF1589) overnight at 4 °C. Rabbit and goat isotype control antibodies were used at similar concentrations (BioLegend, Cat No. 910801 and R&D Systems, Cat No. AB-108-C, respectively), and no significant nonspecific staining was observed (not shown). The sections were then incubated with the corresponding goat anti-rabbit or rabbit anti-goat biotinylated secondary antibodies (Vector, Cat No. PK-6105 and PK-6101) for 45 min at RT, followed by incubation with VECTASTAIN Elite ABC Reagent (Vector, Cat No. PK-6100) for 30 min at RT. Finally, the sections were stained with DAB-EASY tablets (Acros Organics, Thermo Fisher Scientific, Cat No. AC328005000) until the desired staining intensity was achieved. Samples were counterstained with Methyl Green (Vector, H-3402-500) and mounted using EcoMount (Biocare Medical, Cat No. EM897L).
In vitro fluorescence and immunofluorescenceMouse marrow explant cultures were stained with antibodies against Tsg101 (NOVUS, 4A10), Runx2 (Abcam, 76956), Ki-67 (CST, D3B5) and Rank (Abcam, 13918). Mouse Alexa Fluor 555 (CST, 4409) and rabbit Alexa Fluor 647 (CST, 4414) were used as fluorescent secondary antibodies. Osteoclast fusion was evaluated via fluorescence microscopy as previously described38 using phalloidin-Alexa488 and Hoechst (Invitrogen, Cat Nos. H3570 and A30106, respectively) to label the actin cytoskeleton and nuclei. For all the stains, the cells were permeabilized for 10 min in PBS supplemented with 0.1% Triton X-100, and 5% FBS (IF Buffer) was used to suppress nonspecific binding. Then, the cells were incubated with primary antibodies overnight in IF buffer. After washing in IF buffer, the cells were incubated for 2 h at room temperature in IF buffer with secondary antibodies. Images were captured on a Zeiss LSM 800 Airyscan confocal microscope using a C-Apochromat 63x/1.2 water immersion objective.
In situ hybridizationFor human SOST mRNA hybridization, sections were deparaffinized and incubated with RNAscope Hydrogen Peroxide (Advanced Cell Diagnostics [ACD], Cat No. 322335) for 10 min at RT. Target retrieval was performed using ACD Custom Pretreatment Reagent (ACD, Cat No. 300040) for 45 min at 40 °C. Probes were hybridized as instructed and consisted of dapB (bacterial gene, negative control), PPIB (housekeeping gene), and SOST (sclerostin). The remaining hybridization steps were performed using RNAscope 2.5 HD Detection Reagents- RED (ACD, Cat No. 322360) with the exception of AMP5, which was incubated for 60 min. Sections were counterstained with 50% Gil’s Hematoxylin I (Sigma‒Aldrich, Cat No. GHS132) and mounted with EcoMount (Biocare Medical, Cat No. EM897L).
In situ hybridization of Gαs p.R201 variant mRNAs was performed using the novel Advanced Cell Diagnostics [ACD] Basescope duplex system with custom-made probes following the manufacturer’s protocols. For optimization and validation of the method, human bone marrow stromal cell (BMSC) primary cultures containing GNAS p.R201C, GNAS pR201H and WT variants were cultured, pelleted by centrifugation and embedded in OCT cryosectioning media. Five-micron sections were assessed for RNA hybridization of two candidate probes for each GNAS p.R201C, GNAS pR201H and WT variant in collaboration with the ACD team. Images of hybridization with the probes validated for further studies are shown in Fig. S4a.
Briefly, human sections were incubated with RNAscope Hydrogen Peroxide (ACD, Cat No. 322335) for 10 min at RT and retrieved with ACD Custom Pretreatment Reagent (ACD, Cat No. 300040) for 60 min at 40 °C. The positive controls used were PPIB (high-expression housekeeping gene) and POLR2A (low-expression housekeeping gene), and the negative controls used were dapB (bacterial gene) probes and custom-designed probes for wild-type GNAS (ACD, Cat No. 1061061-C1), as well as the two most common mutations in FD: GNASR201C (c.601 C > T, Cat No. 1061041-C2) and GNASR201H (c.602 G > A, ACD, Cat No. 1061051-C2). With the BaseScope Duplex Detection Reagent Kit (ACD, Cat No. 323810), the AMP7 and AMP11 concentrations were increased at 45 min and 60 min, respectively, to increase the staining intensity. Sections were counterstained using 50% Gil’s hematoxylin I and mounted with VectaMount Permanent Mounting Medium (Vector, Cat No. H-5000). The results of staining with the positive and negative control probes are shown in Fig. S4b-g.
Microscopy imagingChromogenically stained PE sections were scanned using a NanoZoomer S60 Digital slide scanner (Hamamatsu, Cat No. C13210-01) at 400 × magnification.
Fluorescent cryogenic sections were scanned using an Axioscan 7 fluorescence scanner (Zeiss, Oberkochen, Germany)37 with appropriate filters for the detection of DAPI (Em 460 nm, Ab 350 nm), Elf97 TRAP (Em 550 nm, Ab 375 nm) and ALP (Em 605 nm, Ab 545 nm). The scanned slides were manually aligned and converted to Adobe Photoshop.psd files.
Cell immunofluorescence images were captured on a Zeiss LSM 800 Airyscan confocal microscope using a C-Apochromat 63x/1.2 water immersion objective. For the osteoclast fusion assay, 8 randomly selected fields of view were imaged using Alexa 488, Hoechst and phase contrast compatible filter sets (BioTek) on a Lionheart FX microscope using a 10x/0.3 NA Plan Fluorite WD objective lens (BioTek) with Gen3.10 software (BioTek).
Microscopy quantificationFor bone content and cellularity, images from H&E-stained PE sections were analyzed using a custom script in ImageJ (NIH). For human sections, complete biopsy sections were analyzed. For mice, 1–3 fields of 0.6 by 0.4 mm of randomly selected FD lesion tissue were analyzed per sample. Nonbone or non-FD tissue was removed using Adobe Photoshop 2021 when necessary (Adobe, San Jose, CA). On ImageJ, pixel-to-area calibration was performed, mineralized and fibrous tissue areas were manually traced, and nuclei were selected in these areas using a semiautomatic color threshold selection followed by a watershed separation plugin. The number of nuclei and the mineralized and fibrotic areas were calculated using the “analyze particles” function. The total number of nuclei obtained with this method was used as the denominator to calculate the ratio of cells positive for TRAP activity, Rank, and Mcm2 in consecutive sections from the human biopsies.
For mouse TRAP and ALP analysis of mouse FD cryosections, multiplexed fluorescence 1 mm × 1 mm images of the distal tibiae were opened in Adobe Photoshop, and the color levels were adjusted and exported as single-layer tiff files corresponding to TRAP, ALP or DAPI for image quantification. The images were subsequently analyzed with QuPath version 0.3.2, an open-source software for digital pathology analysis.39 After calibration of the pixel-to-area ratio of the images, cell nuclei were identified by DAPI staining and counted using the “cell detection” function. TRAP- and ALP-stained areas were traced using the “pixel classification” function, in which the area of fluorescence (µm2) was calculated based on a pixel color intensity threshold. The average TRAP and ALP staining intensity per cell was calculated as the ratio of the µm2 TRAP- or ALP-positive area to the number of nuclei in each quantified image.
We used the QuPath tool in PE scans to manually label and count cells positive for TRAP, Rank (multinuclear osteoclasts or mononuclear precursors), Mcm2 and sclerostin-stained (either by RNA hybridization in humans or immunohistochemistry in mice) and unstained osteocytes. For ACD Basescope™ quantification, we used this tool on 400x images to label hematoxylin-stained nuclei in proximity to the detected GαsR201C or GαsR201H mRNA molecules (red dots), quantified as mutant cells, regardless of the presence of neighboring wild-type Gαs mRNA molecules (green dots). Nuclei associated with only wild-type Gαs mRNA molecules were quantified as wild-type cells. The positive controls PPIB (a high-expression housekeeping gene) and POLR2A (a low-expression housekeeping gene) and the negative control dapB (a characteristic bacterial gene) were used.
Osteoclast fusion efficiency was evaluated by the number of fusion events between osteoclasts with obvious ruffled borders and ≥3 nuclei in phalloidin-Alexa 488/Hoechst images, as described previously.40 Since, regardless of the sequence of fusion events, the number of cell-to-cell fusion events required to generate a syncytium with N nuclei is always equal to N-1, we calculated the fusion number index as Σ (Ni − 1) = Ntotal − Nsyn, where Ni = the number of nuclei in individual syncytia and Nsyn = the total number of syncytia.
RUNX2 and OCN staining in human samples was quantified by a scoring method since the variable intensity of staining made it impossible to quantify based on a cell positivity color threshold. For the secreted factor OCN, the stain was normally dispersed and not associated with individual cells; therefore, a collection of 100x-blinded microscopy images was studied and assigned to 5 scores based on their staining level (0, no stain; 5, maximum stain). Two images of each staining level were subsequently provided to the readers for training purposes. For RUNX2, descriptive scores were developed and are shown in Table S2. All assessments that required human evaluation were examined by at least 3 trained readers in a blinded fashion.
In vitro bone resorption assayBone resorption was evaluated using a bone resorption assay kit from Cosmo Bio (USA) according to the manufacturer’s instructions. In brief, fluoresceinamine-labeled chondroitin sulfate was used to label 24-well, calcium phosphate-coated plates. Media were collected at 4-5 days post-induction, and the fluorescence of the media was evaluated as recommended by the manufacturer.
Cultured cell cytometry analysesBrdU incorporation was used to evaluate the fraction of cells undergoing DNA replication as a surrogate for cell proliferation using a FITC BrdU Flow Kit (BD Pharmingen) according to the manufacturer’s instructions, with one exception. To avoid interference due to GFP expression in combination with GαsR201C, we utilized an α-BrdU-Alexa 555 antibody. Briefly, BrdU was added to media for 4 hours in uninduced cultures, and cultures induced with doxycycline for 4 days that were treated with αRANKL or an isotype control antibody. We also induced cultures that were not labeled with BrdU to act as controls for both autofluorescence and nonspecific antibody labeling. The cells were subsequently digested with trypsin, fixed, and cryogenically preserved until staining and analysis. Approximately 5 000 gated cells were evaluated for BrdU incorporation per condition per replicate. The cells were analyzed using a NovaCyte flow cytometer (ACEA Biosciences, Inc.) with NovoExpress software (v1.5.0) at a rate of <150 events/second.
Culture media determinationsEx vivo cultures were cultured in complete culture media supplemented with FBS depleted of EVs (via ultracentrifugation at 150 000 × g for >2 hours). Twenty-four hours later, the conditioned media was collected, and the cells/large cell debris were depleted via centrifugation (15 min at 4 000 × g).
Enrichment and quantification of extracellular vesicle (EV) fractionsThe EV fraction was enriched via ultracentrifugation (150 000 x g for 1.5 hours). Alternatively, an exoEasy Maxi Kit (Qiagen, Hilden Germany) was used to isolate EVs. EV-enriched fractions were evaluated via Western blotting using anti-Tsg101 (Cat No. 4A10; NOVUS, Louis, MO) and anti-RANK (Cat No. 13918; Abcam, Cambridge, UK) antibodies. Tsg101 and Rank band staining were quantified via densitometry via ImageJ.
Quantification of released factors in culture mediaThe levels of RANKL, M-CSF, SEMA3A, EFNB2 and FAS-L (undetectable) were determined in culture media with the following ELISA kits per the manufacturer’s instructions: MTR00 Mouse TRANCE/RANKL/TNFSF11 Quantikine ELISA Kit, MMC00B Mouse M-CSF Quantikine ELISA Kit, MFL00 Mouse Fas Ligand/TNFSF6 Quantikine ELISA Kit (R&D Systems), LS-F33608 Mouse SEMA3A ELISA Kit, and LS-F6974 Mouse EFNB2 ELISA Kit (LSBio LifeSpan Biosciences, Inc.).
Statistical analysesThe data are expressed as individual data points and means ± SEMs for all values. The results were tested for normality using the Shapiro‒Wilk test, and analyses were performed using nonpaired t tests or Mann‒Whitney tests. For longitudinal measures, paired two-tailed t tests were used to compare baseline values to values at different time points. The data are reported as the average and standard deviation when shown, unless otherwise indicated, and analyses were conducted on GraphPad Prism 8.0.2 by LFdC. Culture data were evaluated by two-tailed t tests paired with donor mouse data and paired t tests for MC3T3-E1 experimental data from JW.
Study approvalThe clinical trial NCT03571191 was approved by the NIH Investigational Review Board, and informed consent was obtained from all the subjects. The study was monitored by a data safety and monitoring committee organized by the National Institute of Dental and Craniofacial Research. Mouse experiments were conducted under a protocol approved by the NIH/NIDCR Animal Care and Use Committee (ASP 19-897).
留言 (0)