Derivation of the dataset
Data from five clinical studies were included in the PopPK analysis dataset, with 370 adult and pediatric individuals receiving balovaptan at repeated doses between 1.5 and 52 mg once-daily (QD). The majority of individuals (n = 321) were from the phase II ASD clinical trials VANILLA in adults (NCT01793441; n = 146) [23] and aV1ation in children and adolescents from 5 to 17 years of age (NCT02901431; n = 169) [24], with doses ranging from 1.5 to 10 mg QD, and relatively sparse PK sampling, mainly at steady state. These phase II study data were enriched with data from three phase I balovaptan PK studies in neurotypical adult participants, with doses ranging from 5 to 52 mg and rich PK sampling from day 1 to steady state. This combination of phase II and phase I data was considered sufficiently informative to describe and estimate non-linearity using a parsimonious approach. The studies included in the PopPK dataset are described below:
Study NCT01418963 (phase I; neurotypical adults) was a single-center, randomized, double-blind, placebo-controlled, first-in-human study to investigate the safety, tolerability and PK of balovaptan [26]. It consisted of three parts: a single-ascending dose study, a multiple-ascending dose (MAD) study and a food effect study. Only data from the 24 participants in the MAD study were included in the modeling dataset. Participants received balovaptan 12, 20, 40 or 52 mg QD (fed) in cohorts of six per dose level for a period of 14 days. Blood samples for balovaptan determination were obtained pre-dose and through 24 h post-dose on day 1, and pre-dose and through 120 h post-dose on day 14, with pre-dose samples taken on each intermediate day.
Studies NCT03579719 and NCT03586726 (both phase I; neurotypical adults) were single-center, non-randomized, open-label, one-sequence, two-period, within-subject studies with similar designs that investigated the effect of multiple-dose itraconazole and rifampicin, respectively, on the PK of multiple-dose balovaptan [27]. Only data from the control period of each study (Period 1), where balovaptan was dosed without the concomitant agent, were included in the modeling dataset. In Period 1, participants received oral balovaptan at a dose of 5 mg QD (NCT03579719; fed) or 10 mg QD (NCT03586726; fasted) for 10 days. Blood samples for balovaptan determination were obtained pre-dose at five time points up to day 10, and at fixed times up to 24 h post-dose on day 10.
Study NCT01793441 (VANILLA; phase II; adults with ASD) was a multicenter, randomized, placebo-controlled, double-blind, 12-week parallel group proof-of-concept study to investigate the efficacy and safety of balovaptan in adult men with moderate/severe ASD and an intelligence quotient (IQ) ≥ 70 [23]. Balovaptan was administered with food at doses of 1.5, 4 or 10 mg QD. Blood samples for balovaptan determination were collected pre-dose and at 2, 4 and 6 h post-dose on days 1, 14 and 84.
Study NCT02901431 (aV1ation; phase II; children and adolescents with ASD) was a multicenter, randomized, placebo-controlled, double-blind study to investigate the efficacy and safety of balovaptan in children and adolescents with ASD and an IQ ≥ 70 [24]. The study comprised an initial PK study to confirm the estimated age-adjusted balovaptan doses yielding blood systemic exposure equivalent to adult 4 mg QD and 10 mg QD, followed by a 24-week parallel two-group trial of placebo versus age-adjusted dosing equivalent to these two adult doses. Recruitment to the 4 mg QD-equivalent arm was subsequently halted early following an interim analysis, while recruitment continued into the 10 mg QD-equivalent arm. Balovaptan doses received across the various age groups over the course of both study parts were 1.5, 2, 3, 4, 5, 7 and 10 mg QD, and balovaptan was taken with or without food. Blood samples for balovaptan determination were collected in the PK cohorts at 4 h post-dose on day 1, pre-dose at day 10 in a subset of participants, and pre-dose and up to 6 h post-dose at week 2. In addition, for all patients (including the PK cohorts), samples were taken at week 8 at one post-dose time point in the evening (at home); and at weeks 12 and 24 pre-dose, 2 h post-dose and at the end of the clinic visit (≥ 4 h post-dose).
PopPK model development
Model building was conducted in NONMEM v7.4.3 (ICON Development Solutions, Ellicott City, MD, USA) using first-order conditional estimation with interaction (FOCE-I). Data processing and post-processing of NONMEM analysis results was carried out in R v3.5.2 (Comprehensive R Network: https://cran.r-project.org/).
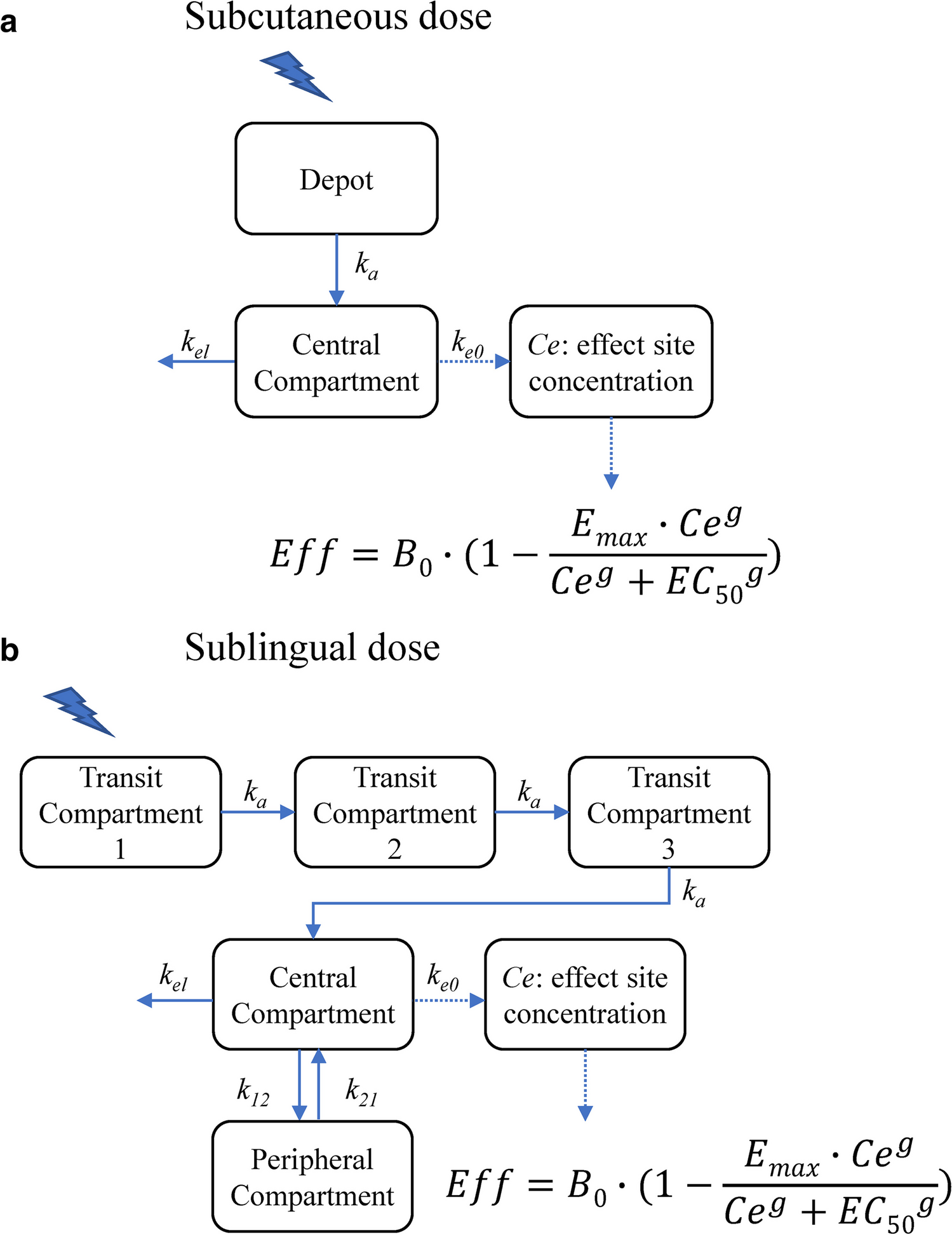
One- and two-compartment disposition models with first-order linear absorption and elimination were fitted to the data as a starting point for further development. Allometric scaling of CL/bioavailability (F) and apparent volume of distribution (V/F) was added a priori, evaluating fixed versus estimated allometric exponents, to account for the wide ranges in age and body weights, using a power relationship based on a reference weight of 76 kg. This reference weight was the median weight of the neurotypical adult volunteers in the dataset, which lay between the median weight of the adult ASD trial participants (89 kg) and the pediatric trial participants (56 kg).
Several approaches were tested to address observed non-linear PK behavior. Diagnostic plots from initial modeling steps showed a trend towards a decrease in the apparent central volume of distribution (Vc/F) with increasing dose, indicating that saturable (i.e., capacity-limited) binding might be the underlying process. Two modeling approaches for saturable binding were explored. The first of these was the commonly used explicit binding model that describes a balovaptan mass transfer between the central and a saturable binding compartment. The second, less common approach was an empirical model that introduces a dynamic Vc/F, as a non-linear function of the balovaptan amount in the central compartment (see Supplementary Materials for model equations). Other approaches to model non-linearity included non-linear elimination from the central compartment, non-linear bioavailability, and eventually, an empirical gut extraction component, with a hypothetical turnover compartment that controls the extent of gut extraction in an exposure- and time-dependent manner.
Absorption modeling compared a first-order absorption rate with a transit compartment absorption model (TCAM) [30], with the food effect (fed versus fasted) included in the structural model a priori. Inter-individual variability (IIV) was estimated for CL/F, Vc/F and the absorption parameters (Ka or mean transit time (MTT)) using a diagonal OMEGA matrix. Several residual error models were tested to characterize the residual unexplained variability (RUV), including a proportional model, a combined additive and proportional model, and a time-varying RUV model.
Covariate analyses were undertaken for both body size and age effects on CL/F and V/F. Lean body mass (LBM) was estimated from body weight and height according to the methods described by Boer [31] for male and female adults, and by Peters et al. [32] for pediatric participants. The effect of LBM and of body mass index (BMI) were evaluated as potential alternatives to allometric scaling based on body weight. Age effects were modeled as continuous relationships using a power model or an asymptotic model.
Model selection criteria were based on the maximum likelihood of model fit (objective function value (OFV)), goodness-of-fit plots, physiological plausibility, precision of parameter estimates, model numerical stability and the condition number. The model qualification was based on acceptable visual predictive checks [33]. Confidence intervals on the parameter estimates were generated using a bootstrap approach with 500 replicates.
Pediatric exposure simulations
The final PopPK model was used to simulate alternative age-based balovaptan dosing scenarios in a pediatric and adult population with ASD aged 2 years and above. Age–weight–sex distributions were sampled from a large database comprised of the participants from the aV1ation and VANILLA trials together with 1000 virtual participants aged 2–25 years from a pediatric and adult ASD population that was simulated in Simcyp (Certara, Inc., Princeton, NJ, USA).
Three-thousand individual PK profiles were simulated to derive steady-state area under the curve (AUCss) for each dosing scenario. The target scenario was an AUCss distribution that was homogeneous across the pediatric age range and equivalent to adult exposure at all ages. Based on the results from these simulations, an updated age-based dosing algorithm was proposed for future studies of balovaptan in pediatric participants aged 2 years and above.
IV exposure and RO simulations
Plasma kinetics and brain V1a RO following intravenous (IV) balovaptan administration were simulated to explore dosing strategies for AIS patients in Study NCT05399550. The target for optimal dosing and administration was a RO of at least 80% in at least 95% of the simulated individuals over a period of 72 h, with a subsequently rapid decline in RO thereafter.
Simulations relied on assumptions that were derived from data outside of the indicated administration route and patient group. For the simulation of plasma PK profiles it was assumed that oral CL/F and V/F were identical to IV CL and V, based on the observation of 100% bioavailability in healthy neurotypical adult individuals [26]; IV infusion was therefore modeled as a zero-order input into the central compartment with distribution and elimination unchanged from the oral model.
For the simulation of RO, balovaptan concentration at the brain V1a target site was assumed to be equivalent to the plasma-free fraction, based on negligible P-glycoprotein extraction ratio measured in vitro [22]. In absence of a suitable V1a PET tracer, balovaptan binding affinity was estimated in an ex vivo experiment using human platelet aggregates from whole blood. The results correlated well with the values from the in vitro affinity of balovaptan to the human V1a receptor and from the agonist-mediated calcium fluxes in intact cells expressing human V1a receptors using Fluorescent Imaging Plate Reader (FLIPR). Platelets are known to express the V1a receptor [34, 35] and thus the brain RO was modeled assuming that the brain receptor binding constant is the same as the platelet receptor dissociation constant. Other assumptions underlying the RO predictions include (i) PK in the (elderly) AIS population is not different from the younger population used for developing the dataset, and (ii) cerebrospinal fluid (CSF) concentration is reflective of brain distributions.
Based on these assumptions, the PK pharmacodynamic (PD) relationship for brain RO was simulated as:
$$\mathrm=\frac_\times _}_+ \left(_\times _\right) } \times 100$$
where RO is the percent predicted brain receptor occupancy; Cp is the plasma balovaptan concentration; fu_plasma is the plasma-free balovaptan fraction; and Kb is the dissociation constant.
Several dosing scenarios were simulated in order to achieve the target RO without exceeding the previously observed PK exposure. All scenarios included a high starting dose (50 or 60 mg) to achieve a rapid onset of high RO without exceeding the highest plasma concentrations observed in previous studies. For the follow-up doses, scenarios with either 24- or 36-h dosing intervals were tested, resulting in either a total of three or two doses within the 72-h treatment duration. Follow-up doses between 10 and 30 mg were evaluated. Two-thousand participants were simulated for each scenario, assuming adult “typical” parameters including the 76 kg reference weight of the neurotypical adults in the model dataset. A sensitivity analysis was also performed, assuming a body-weight distribution for a reference population of mean weight 82.2 kg with an SD of 20.3 kg and a range of 41–136 kg. These weight parameters were derived from the first 258 patients screened for entry into the ongoing TIMELESS study (NCT03785678) of tenecteplase thrombolysis immediately (4.5–24 h) after AIS (unpublished data).



留言 (0)