
記住我



The basic information of the 158 subjects is shown in Table 1, and there was no statistically significant difference in sex or age (P > 0.05).
Table 1 Basic information of 158 subjectsHigh-quality sequences and ASVs/OTUsA total of 16,022,648 (reads) high-quality sequences were obtained by using the DADA2 method to remove primers, quality filtering, and removal of chimeras, with an average of 101,409 (reads) high-quality sequences per specimen. Among all the specimens, the maximum number of sequences was 144,325 (reads), and the minimum number of sequences was 48,760 (reads). The average length of each sequence was 424 bp (base pair, bp), and 99.9% of the high-quality sequences ranged from 400 to 431 bp. The high-quality sequences were aggregated into 780 operational taxonomic units (OTUs) representing 387 individual species belonging to 127 genera, 92 families, 58 orders, 39 classes, and 19 phyla. A total of 1,986 OTUs were recorded in all groups, indicating the presence of the core microbiome of the ocular surface (Fig. 1–A, B, C).
Fig. 1
(A) Sequence length distribution; (B) the number of ASVs/OTUs; (C) ASV/OUT Venn diagram; (D) Rarefaction curve; (E) Species accumulation curves
Evaluation of sequencing depth and sample sizeThe shape of the sparsity curve provided insights into the impact of sequencing depth on microbial community diversity. In our study, the sparsity curve demonstrated a gradual plateau as the amount of sequencing data increased, suggesting that the current sequencing depth adequately captured the richness and evenness of microbiome within the sample. The species accumulation curve indicated that the sample size was appropriate for the study (Fig. 1D and E).
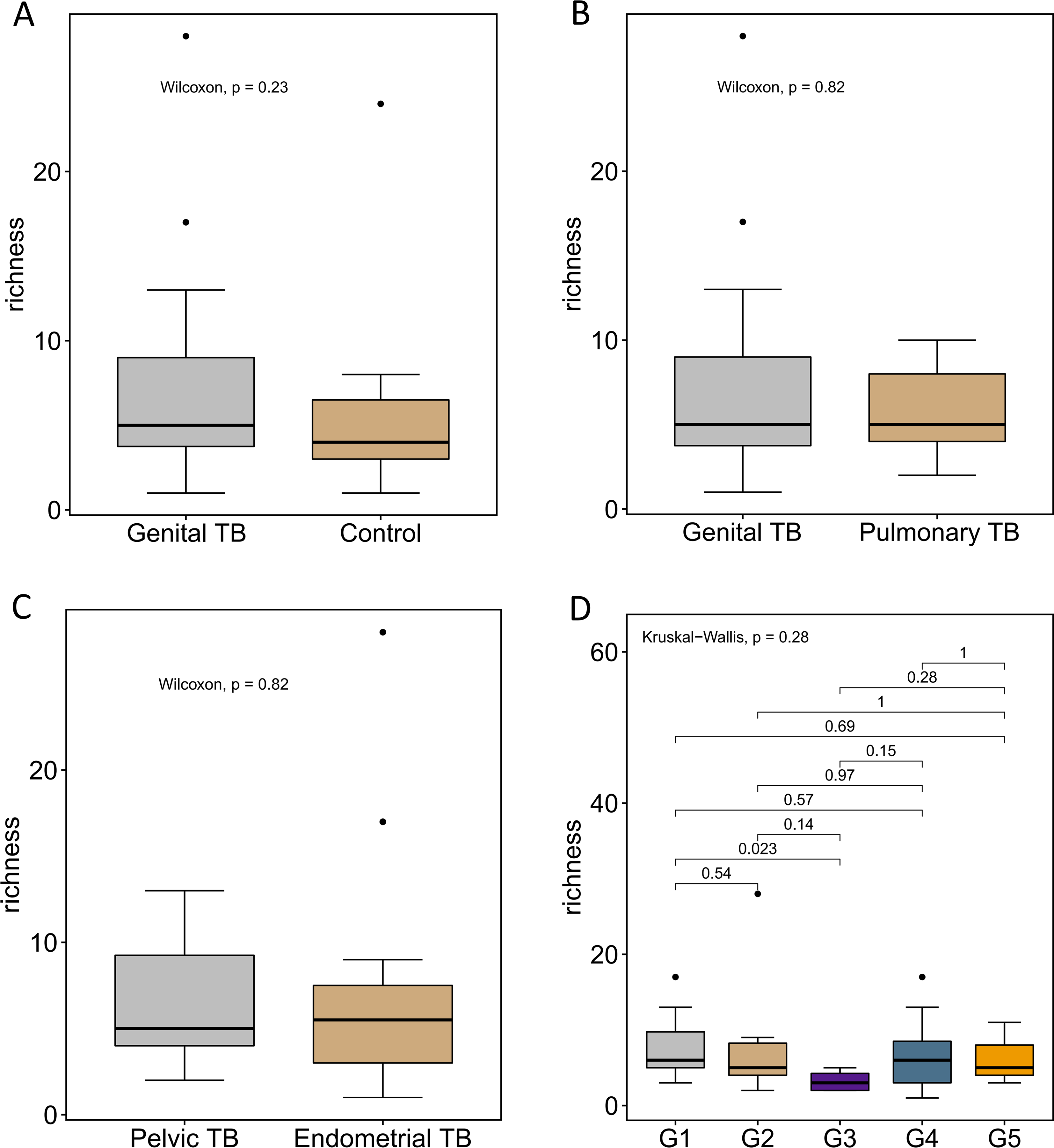
α diversity analysis and β diversity analysisRichness was represented by Chao1 and the observed species index, diversity was represented by the Shannon index, evolution diversity was represented by Faith’s PD index, evenness was represented by Pielou’s evenness index, and the coverage diversity was represented by Good’s coverage. The results of these α diversity indices showed that the ocular surface microbiome in the NoDM-DE group had higher richness, and the ocular surface microbiome in the diabetes group had more diversity and uniformity. The t-test showed significant differences among all groups (Fig. 2A).
A partial least squares discriminant analysis (PLS-DA) model was established using the relative abundance data at the species level. The Bray-Curtis distance algorithm was employed, with a two-dimensional NMDS representation and a 0.95 elliptic confidence. The results of the principal coordinate analysis (PCoA) and NMDS analysis are shown in Fig. 2B and C. The species composition varied significantly between the groups, indicating distinct differences.
Fig. 2
(A) α-Diversity index analysis of the four groups; (B) PCoA analysis; (C) NMDS analysis; (D) the relative abundance of four groups at phyla level (top 10); (E) the relative abundance of four groups at genus level (Top 20); F Species composition heatmap at the genus level (Top 20)
Microbiological taxonomic analysisThe bacterial composition of the ocular surface in the four groups was compared, and the 16 S rRNA sequences of individual bacteria were classified at the phylum and genus levels. At the phylum level (Fig. 2D), the 16 S rRNA gene sequencing of ocular surface bacteria in the four groups revealed the presence of four significant phyla: Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. The abundance of Proteobacteria in the DM-DE group (58.00%) was higher compared to the control group (53.98%). In the DM-DE group, the abundance of Firmicutes (26.66%) was significantly higher than that in the other groups (DM-NoDE group: 19.40%, NoDM-DE group: 21.16%, control group: 21.44%). The control group exhibited the highest abundance of Actinobacteria (22.78%) compared to the DM-DE group (13.30%), DM-NoDE group (12.10%), and NoDM-DE group (14.49%). Bacteroidetes exhibited the highest abundance in the DM-NoDE group (2.58%) compared to the DM-DE group (1.03%), NoDM-DE group (1.06%), and control group (0.95%).
At the genus level, the majority of the 16 S rRNA gene sequencing results from the four groups of ocular surface bacteria were assigned to 20 genera (Fig. 2E). These genera included Pseudomonas, Anoxybacillus, Corynebacterium, Cupriavidus, Chelatococcus, Curvibacter, Ochrobactrum, Streptococcus, Enhydrobacter, Lactobacillus, Staphylococcus, Coprococcus, Novosphingobium, Blautia, Agrobacterium, Rhodococcus, Bacteroides, Finegoldia, Nesterenkonia, and Acinetobacter. In the DM-DE group, Anoxybacillus, Cupriavidus, and Chelatococcus were significantly more abundant compared to the other groups, while Pseudomonas exhibited significantly lower abundance than in the other groups. The control group had the highest abundance of Corynebacterium (Fig. 2F).
In all samples, the microbiome with abundances greater than 1% were clustered at the phylum and genus levels, among which, Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes were the dominant bacterial phyla, and Pseudomonas, Anoxybacillus, Corynebacterium, Cupriavidus, Chelatococcus, and Curvibacter were the dominant bacterial genera.
Analysis of differences between groupsLEfSe analysis in this study showed that abundance of Firmicutes in DM-DE group was significantly higher than that in other groups (P < 0.05); the abundance of Bacteroidetes in DM-NoDE group was significantly higher than that in other groups (P < 0.05). The abundance Actinobacteria in Control group was significantly higher than that in other groups (P < 0.05); The abundance of Anoxybacillus and Chelatococcus in DM-DE group were significantly higher than those in other groups (P < 0.05) > The abundance of Pseudomonas in DM-NoDE group was significantly higher than that in other groups (P < 0.05); and the abundance of Staphylococcus and Clostridium in NoDM-DE group were significantly higher than those in other groups (P < 0.05). The abundance of Corynebacterium in the control group was significantly higher than that in the other groups (P < 0.05) (Fig. 3A).
Fig. 3
(A) LEfSe analysis in four groups; (B) analysis of differences between the four groups; (C) PCA analysis
Using the Bray-Curtis distance matrix file and the python scikit-bio package (permanova), the difference analysis showed that there were statistically significant differences in species abundance between the DM-DE group and other groups in their conjunctival swabs (P < 0.05) (Table 2; Fig. 3B and C).
Table 2 Analysis of differences between the four groups
留言 (0)