Complete congenital sternal cleft (CS), also called absent sternum, is a very rare and potentially life-threatening birth defect [1]. It results from failure of the process of midline mesenchymal strip fusion during embryonic development [2]. It accounts for only 0.15% of all chest wall malformations. It can be complete and incomplete type.
Complete CS is very rare and involves splitting of the sternum from the manubrium to the xiphoid. This variety is also strongly associated with complex intracardiac defects such as TOF, Cantrell’s pentalogy, ectopia cordis, and many others. Noncardiac anomalies may be cervicofacial hemangiomas, diastasis recti, and cleft mandible. Incomplete CS presents with chondral bridges either in the upper part (inferior-type incomplete CS) or in the lower part (superior-type incomplete CS) of the sternum. It is very often associated with vascular dysplasias. A supraumbilical raphe is present in 30% of cases of CS. The etiology of CS is speculative.
There are two theories involved:
•Failure of fusion of lateral sternal bands with midline mesodermal structures between the 6th and 9th gestational weeks.
•Early amnion rupture may result in multiple defects by pressure necrosis, incomplete embryogenesis, and tearing and tethering by amnion bands. Surgery is advocated to protect the underlying heart from external trauma to allay parental anxiety and to prevent recurrent respiratory tract infections [3].
CS might occur in isolation or along with defects of abdominal wall, diaphragm, pericardium, and heart. These patients are at increased risk of mediastinal trauma, hypothermia, increased insensible fluid losses, cyanosis, and recurrent infections of the chest [2].
Published experience with absent sternum is limited to sparse case reports, and no preferred method for management has been described [1].
Early diagnosis and surgical correction give the infant the best chance of survival [4]. Isolated CS is best repaired primarily in the neonatal period because extreme compliance and pliability of bony thorax permit direct sternal closure. However, the presence of complex cardiac defects postpones the sternal repair until the age and weight of elective operation of the cardiac malformation are reached [3, 5]. Reconstructive surgery of absent sternum should be performed by primary closure using combined periosteal advancement flap and sliding osteochondroplasty during the neonatal period when the chest wall is highly compliant and closure can be achieved without significant cardiopulmonary compromise [2].
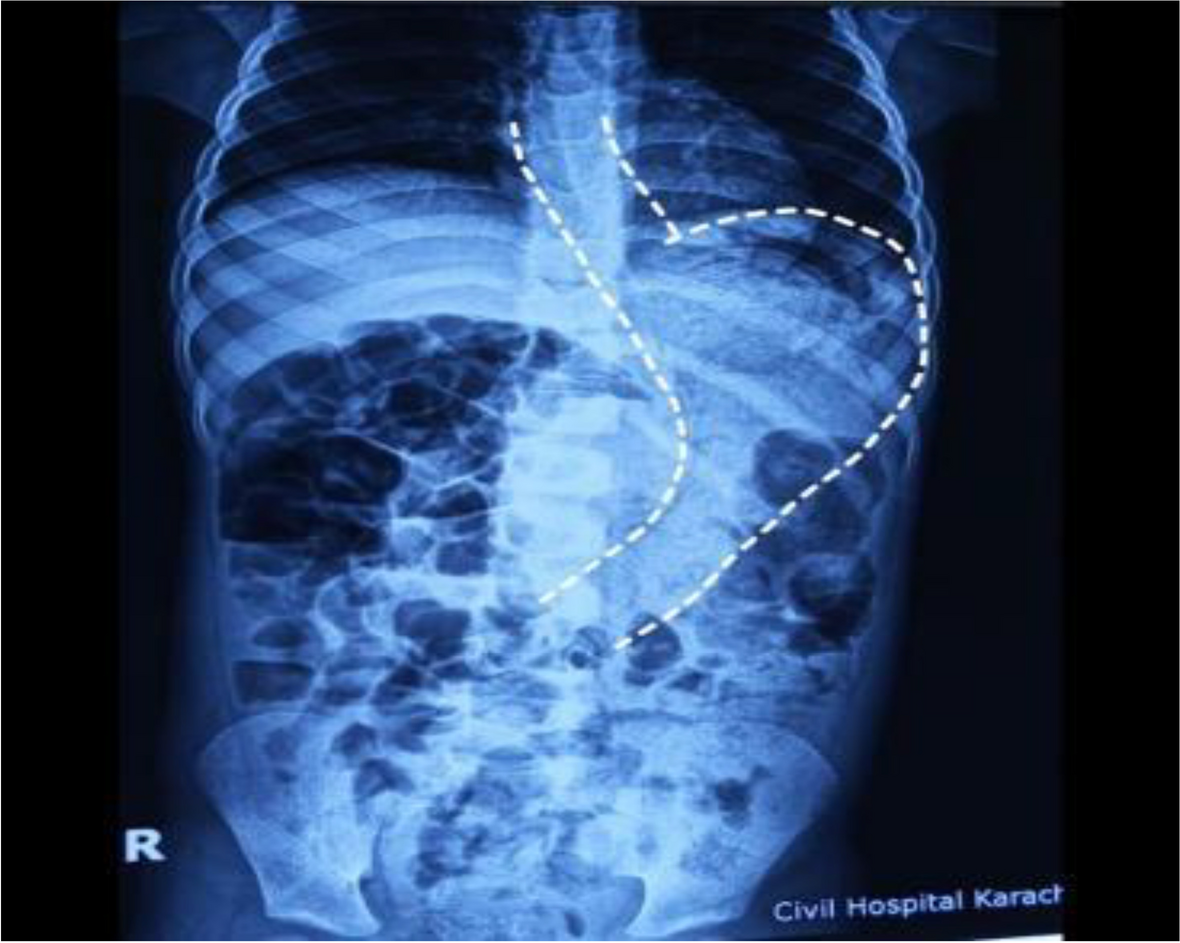
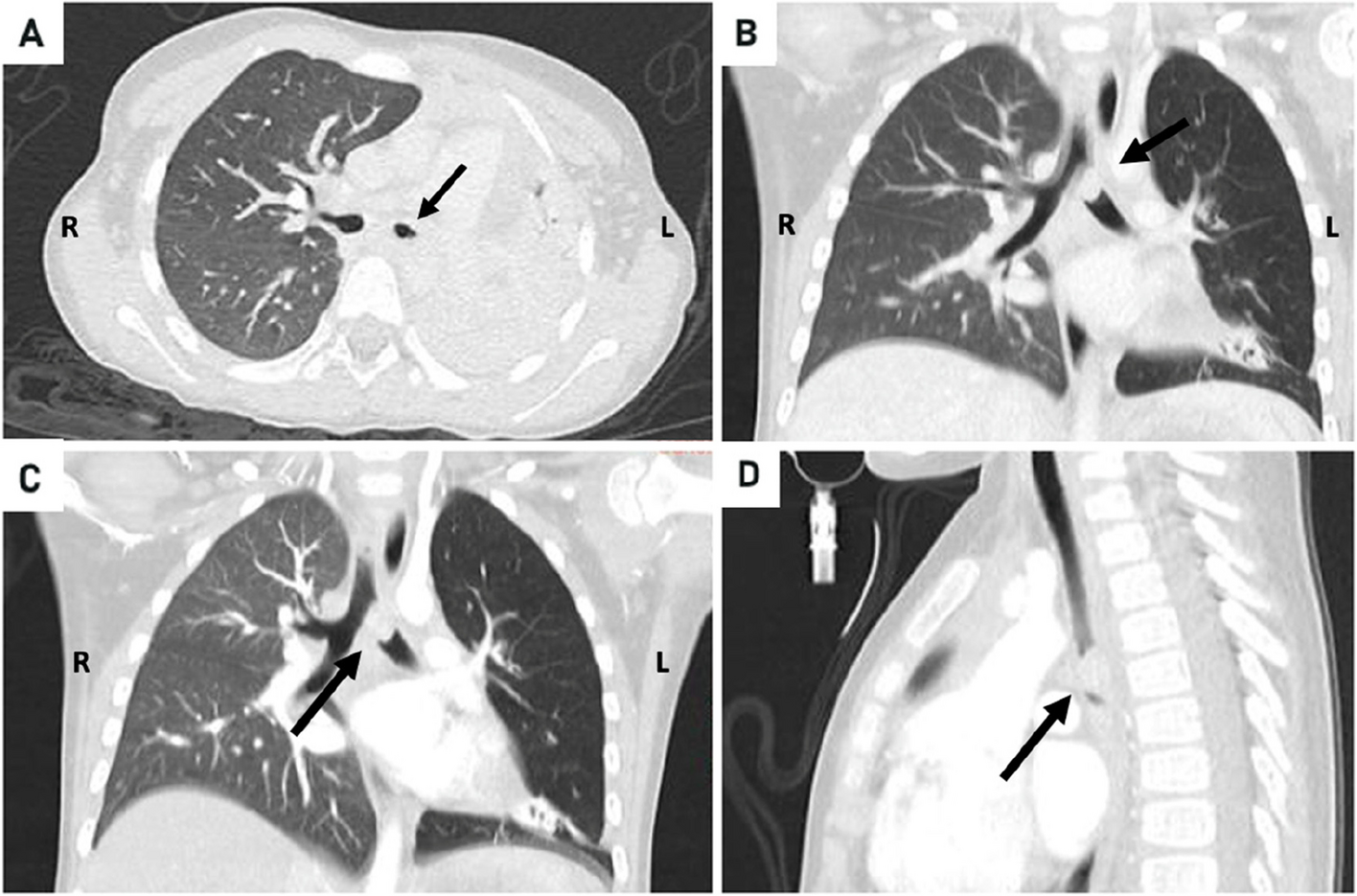
In our case, combination of complete sternal cleft associated with intracardiac defects like tetralogy of Fallot with vascular anomaly like right aortic arch and bilateral vena cava obviously makes it extremely rare.



留言 (0)