

記住我
Cancer is one of the highest causes of morbidity globally (1). Accumulation of genetic mutations brings about this disease; however, the progression would not have been possible by itself alone, but rather with the help of a multitude of factors constituted in the tumor microenvironment (TME). The heterogeneous cellular and non-cellular elements of the TME, such as immune cells, cancer-associated fibroblasts (CAFs), matrix components, and chemokines, operate either against or with the disease (2). To combat transformed cells, known as the elimination phase, inflammation in the TME later turns to an equilibrium phase, with the coexistence of surviving cancer cells along with inflammatory subsets (3). Eventually, chronic inflammation ends up helping the tumor (escape phase) (3), pointing towards the two hallmarks of cancer, namely, tumor-promoting inflammation and avoiding immune destruction (4). Cancer, being a master manipulator, evades the anti-tumor immune surveillance via escaping detection, inducing death, or changing it to a phenotype that serves its purpose (5, 6).
Tumor-associated macrophages (TAMs) occupying more than 50% of tumor-infiltrating cells can be broadly classified as M1-like, pro-inflammatory subtype and M2-like, immunosuppressive subtype (3, 7). During the early stages of cancer detection by immune cells, tissue-resident macrophages (TRMs) attack them by developing pro-inflammatory conditions and presenting phagocytosed antigens on their major histocompatibility complex (MHC) class II receptor (8, 9).
As a result, an adaptive immune response is generated with the invasion of anti-tumor M1 macrophages, CD8+ T cells, and natural killer (NK) cells, which maintains the inflammatory conditions to challenge cancer cells while also constantly replenishing their population (9, 10). Because cancer has an immunomodulatory feature, this neoplastic fraction develops ways to prevent phagocytosis, elude detection, recruit pro-tumor M2 macrophages, and, most intriguingly, produce cytokines to shift the M1 to the M2 type (11). As a result, what started as an effective tumor-eliminating response ends up with a tumor-promoting one, exacerbating the disease. When discussing tumor elimination, it is appropriate to emphasize the tiny subpopulation known as cancer stem cells (CSCs), which, according to mounting evidence, is the “root cause” of therapeutic failures, relapse, tumor progression, invasion, metastasis, and even cancer initiation (12, 13). CSCs employ diverse modalities and hide within the cancer niche to protect themselves from stress signals such as chemotherapeutic drugs and immune attacks (12). Understanding the CSC–TAM crosstalk is therefore critical to get the big picture.
In this review, we will address the important role that TAMs play in TME, whether to suppress or aid cancer. We will have a better understanding of the many underlying processes of the bidirectional interaction between cancer and TAMs, as well as that of CSC–TAM, which not only shapes the tumor landscape but also defines the immunological environment, as well as abetting disease progression and therapy failure. Understanding this crosstalk would aid us in developing strategies to effectively target cancer with its supporting immune arm.
2 Tumor microenvironmentThe tumor body is not just a collection of cancer cells but also contains a variety of resident and infiltrating host cells (14). Tumor cells constantly compel the TME to undergo extensive molecular, cellular, and physical alterations to sustain their growth and development. The composition of a developing TME is a complex, dynamic entity, which varies according to the stages and kind of tumor with distinctive features including immune cells, stromal cells, blood vessels, and extracellular matrix (ECM). Although various adaptive and innate immune cells that promote or inhibit tumor growth infiltrate the tumor site, it is shown that TME actively fosters cancer development rather than simply acting as a silent observer (15). Tumor growth and its constantly accumulating mutations change the TME immunophenotype from immunogenic to tolerogenic, reprogramming pro-inflammatory immune cells to behave in favor of tumor growth, provide resistance to applied therapies, and fail to perform their intended function (10).
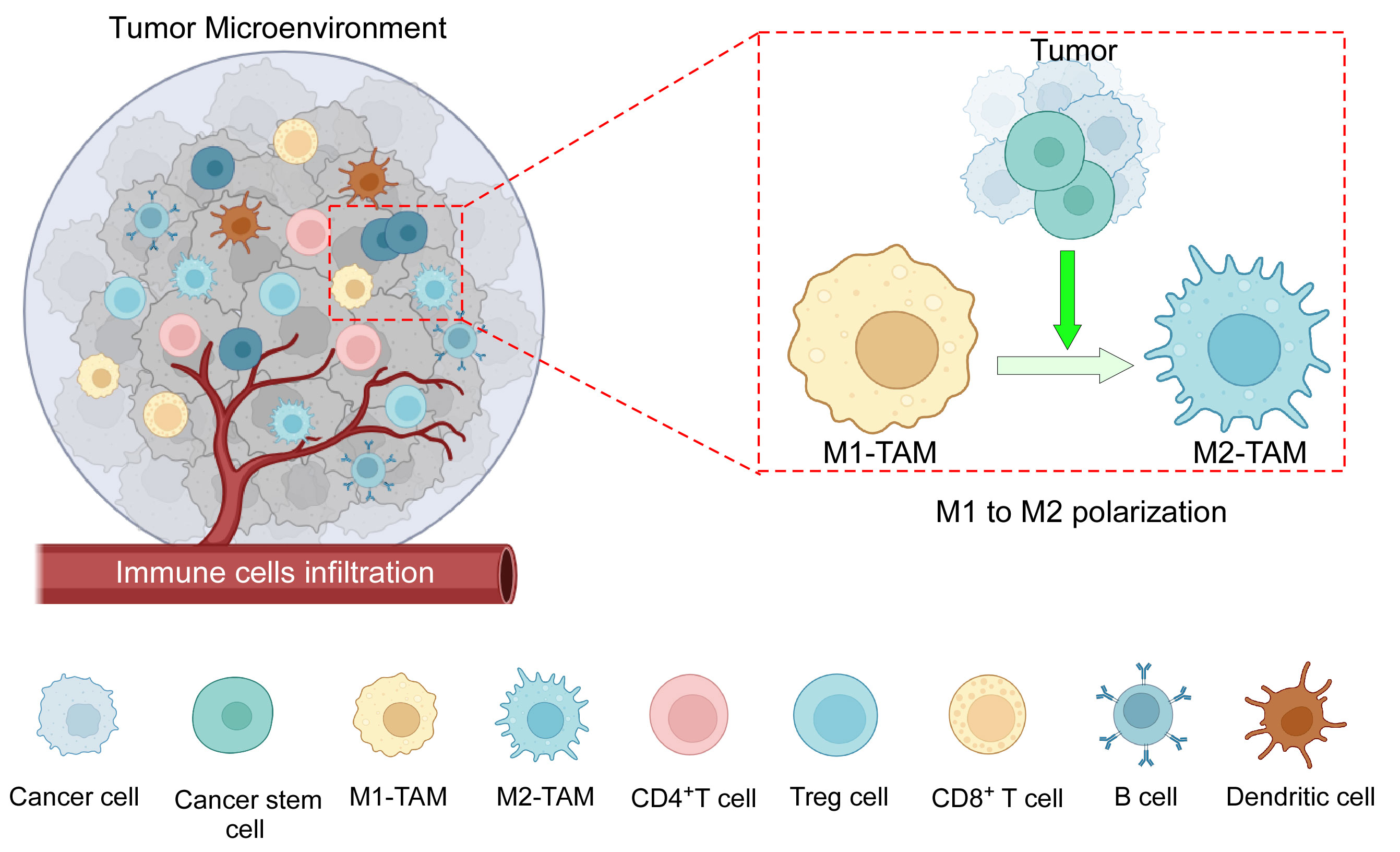
TME immune components include both pro- and anti-inflammatory cells of innate and adaptive immunity (Figure 1). Macrophages are particularly significant because they influence both innate and adaptive immune responses (16). TAMs in tissues were assumed to originate solely from bone marrow (BM)-derived monocytes. Recent evidence suggests the existence of TRMs, which evolve from embryonic progenitors and can survive without the assistance of BM-derived monocytes (17, 18). It is recognized that during the early stages of cancer, only embryonic lineage-derived TRMs aid tumor progression, and that eventually, in response to signals from developing tumors, blood-derived monocytes infiltrate and further enhance the process (17). In the murine mammary carcinoma model, TRMs decrease in number whereas BM-derived monocytes increase with the advancement of cancer (19). However, in tumors such as pancreatic ductal adenocarcinoma, both types of TAMs are present at the same time, with TRMs secreting significant levels of fibrotic proteins and infiltrating macrophages primarily serving as antigen presenters (18). Furthermore, TRMs in ovarian cancer are thought to induce metastasis and maintain the CSC population (20). TRMs are also found to be the most common type of macrophage associated with brain tumors (21), and they have been linked to glioma formation and progression (22). In TME, when considering the overall TAM population, they can be broadly subtyped as anti-tumor M1 and pro-tumor M2 phenotypes. Anti-tumor M1 macrophage demonstrates tumoricidal activity, whereas immunosuppressive M2-polarized macrophages, commonly termed as M2-TAMs, are the most prominent immune cell population within the TME (9). Incidentally, infiltrating macrophages in the majority of tumors are also predominantly of the pro-tumor M2 phenotype. TAMs have a range of roles depending on tumor type, but they are generally important in all stages of carcinogenesis, from tumor initiation to secondary tumor progression. They contribute to angiogenesis, lymphangiogenesis, immunological suppression, hypoxia-induced tumorigenesis, metastasis, and drug resistance, among others (7). TAMs and CSCs have recently been discovered to communicate with each other (23). TAMs are emerging as a critical diagnostic indicator due to the frequent association between their abundance and a poor prognosis (24). As a result, it is critical to investigate TME-induced macrophage skewing and TAM-related pro-tumorigenic effects.
Figure 1 The components of the TME: TME consists of an intricate interplay among various cellular components, including tumor cells, CSCs, tumor-associated macrophages (M1 and M2), CD4+ and CD8+ T cells, B cells, dendritic cells, immune-suppressive Treg cells, as well as other cells, and a complex network of dysregulated vasculature. In addition to several mechanisms, cancer cells along with CSCs play an active role in the polarization of pro-inflammatory M1 macrophages towards anti-inflammatory M2 macrophages. Created with BioRender.com.
3 Types of macrophagesMultiple macrophage phenotypes have been identified so far based on diverse surface receptor expressions, secretory patterns, and activities (25). The multiple unique markers that macrophages express on their membranes result in a range of phenotypes dependent on TME signals, leading to a high degree of plasticity. While TAMs are closely associated with the tumor, cancer-associated macrophage-like (CAML) cells are disseminated tumor cells found in peripheral blood (26). While differentiated macrophages are a rare phenomenon as a circulating population of cells, CAML is defined as macrophages with phagocytosed tumor fraction, has been studied in breast, pancreatic, and prostate cancer, and may serve as a biomarker for these cancers (27). CAMLs are more commonly found in people with advanced malignancies than in those with benign tumors. Surprisingly, the number of CAMLs increases following chemotherapy, which may be related to the efficiency of the treatment because increased phagocytosing macrophages are proportionate to dying cancer cells (26, 27). Augustyn et al., on the other hand, used data from a phase 2 clinical trial (NCT02525757) to show that the existence of giant CAMLs was associated with metastases and poor survival despite immunotherapy and chemoradiation (28). Furthermore, patients with programmed cell death ligand 1 (PDL1) expressing CAML when treated with immunological checkpoint inhibitors (ICIs) had significantly greater overall survival than those who were not treated with ICIs in metastatic lung cancer (29). In general, TAMs are classified as either traditionally activated anti-tumor M1 or alternatively activated pro-tumor M2 phenotypes. According to in vivo wound healing studies, the early stages of wound healing are characterized by the activation of pro-inflammatory M1-like macrophages, which gradually give way to an anti-inflammatory M2-like macrophage phenotype (30). The macrophage colony-stimulating factor-1 receptor (CSF1R) is the primary lineage regulator of virtually all macrophages, regardless of origin. This class III transmembrane tyrosine kinase receptor is expressed by the majority of mononuclear phagocytic cells (25). IFNγ and IL1β, secreted by Th1 cells and bacterial lipopolysaccharide (LPS), drive macrophages to polarize towards the M1 phenotype, whereas IL4 and IL13, secreted by Th2 cells, cause the M2 phenotype to be dichotomized (31).
Macrophages in the M1 end of the continuum have a pro-inflammatory phenotype and express the surface markers CD86 and CD64 (32); MHC-II and macrophage receptor with collagenous structure (MARCO) (33, 34); nitric oxide synthase-2 (NOS2) and suppressor of cytokine signaling-1 (SOCS1); and pro-inflammatory cytokines (IL6, IL12, IL1β, and TNFα) and chemokine ligands (CCL2, CCL5, CXCL9, CXCL10, and CXCL11) (32). All of these indicators show their strong phagocytic and cytotoxic power, ability to draw T and B cells to the infection site, and prodigious ability to deliver antigens (35). In contrast, pro-tumor M2 macrophages with surface markers CD36, CD206, and CD163 are immunosuppressive and anti-inflammatory, helping with tissue repair, angiogenesis, and phagocytosis to reduce and “clean up” after inflammation. They are also Th2 activators and Th1 inhibitors (25, 35–37). M2-like macrophages are typically characterized by their poor ability to present antigens, having low IL12 and high IL10, IL4, and IL13 secretory profiles (35). M2 macrophages also express/secrete transforming growth factor-beta (TGFβ), peroxisome proliferator-activated receptor-gamma (PPARγ), CCL14, CCL22, and arginase-1 (ARG-1) (38).
M2-like macrophages are more functionally diverse than M1-like macrophages because they have many subtypes (M2a, M2b, M2c, and M2d), each with a distinct combination of cytokine and chemokine profiles (39). M2a macrophages express higher levels of IL10, TGFβ, and the chemokines CCL17, CCL18, CCL22, and CCL24, all of which are linked to Th2-polarized allergic inflammation. IL4 and/or IL13 stimulate the production of M2a macrophages. Immune complexes (ICs), LPS, Toll-like receptors (TLRs), or the IL1 receptor antagonist (IL1ra), on the other hand, sustains M2b macrophages, which are characterized by the production of TNFα, IL1β, IL6, IL10, and CCL1. A TGFβ-, glucocorticoid (GC)-, prostaglandin E2-, and IL10-rich environment induces M2c macrophages, which continue to express IL10 and TGFβ; thus, they are crucial regulators for inflammation resolution and tissue healing. Finally, M2d macrophages have been shown to contribute to angiogenesis by expressing vascular endothelial growth factor (VEGF) and IL10 when stimulated by TLR, adenosine A2A receptor ligands, and IL6 (40, 41).
In the context of cancer, these pro-tumor M2 macrophage subsets share the function of tumor development and immune response suppression via multiple pathways (42). For example, VEGF and CCL18, which are released by M2a macrophages, promote breast cancer cell motility and angiogenesis (43, 44). Furthermore, M2a macrophages contribute to the development of lung cancer cells via the IL4/STAT6 signaling pathway. STAT6-expressing M2a macrophages are required for tumor cell growth (45). IL4, which is released by both tumor cells and M2a macrophages, promotes more macrophages to polarize to the M2a phenotype, resulting in a positive feedback loop (45). M2b macrophages proliferate and replace M1 macrophages as hepatocellular carcinoma (HCC) advances (46). These cells release CCL1 in order to attract Th2 and Treg cells that express CCR8, thereby promoting a pro-tumorigenic environment (47). The CCL1/CCR8 signaling mechanism also enhances tumor cell motility, proliferation, and metastasis (48, 49). Furthermore, M2b macrophages express higher levels of indoleamine 2,3-dioxygenase (IDO), IL10, and IL6, all of which are immunosuppressive factors (50). M2b macrophage-secreted IL10 promotes Treg cell differentiation from naive T cells (42), whereas secreted IL6 activates Th2 cells, which promote tumor progression (51). In breast cancer patients, the percentage of circulating M2c macrophages is associated with a poor prognosis (52). Yuan et al. demonstrated that M2c macrophages promote lung tumor growth (53). Kim et al. (54) revealed evidence that IL10-induced M2c macrophages promote tumor development in mouse melanoma models. Furthermore, both in vitro and in vivo, M2c macrophages enhanced endothelial cell mobility and tube formation, implying that M2c may boost tumor growth through increased angiogenesis (55, 56). M2d macrophages in gastric cancer release a number of pro-tumorigenic molecules, including IL10 and TGFβ, to promote cancer cell proliferation and migration (57). M2d macrophages release VEGF and matrix metalloproteinase 9 (MMP9), which promote ECM breakdown, angiogenesis, and metastasis (58). They also produce IL6 in various cancer. The canonical IL6/JAK/STAT3 pathway is related to survival, angiogenesis, metastasis, proliferation, and drug resistance. M2d macrophages also express immunosuppressive factors such as IDO, IL10, and PDL1 (42, 58).
4 Macrophage polarization states in the TMEM1-like macrophages penetrate the TME during the early phase of tumor by antigen presentation and secreting CXCL9, CXCL10, and CXCL11, which chemoattract CXCR3-expressing effector immune cells like CD8+ cytotoxic T cells and NK cells (9, 59). A pro-inflammatory environment containing IFNγ is associated with the preservation of CXCL10+ M1 macrophages (60, 61). In M1 macrophages, IFNγ activates the STAT1 pathway, which is responsible for the production of CXCL10 (62–64). In ovarian cancer, CXCL10+IRF1+STAT1+ M1 resulted in improved antigen processing capacity and T-cell infiltration, as well as a better patient prognosis (60). Effector and memory T cells unlike naïve T cells express CXCR3 (65). As a result of increased infiltration of effector cells, elevated CXCL10 expression has been associated with patient survival in ovarian cancer (66). CXCR3 inhibition reduced CD8+ T-cell infiltration and accelerated tumor progression in mouse models (67). CXCR3 is shown to be expressed in Tregs in HCC, which can hinder effector immune cells from acting due to competitive recruitment in the TME (68). Furthermore, CXCL10 induces chemotaxis of CXCR3+ NK cells, resulting in tumor regression (69). STAT3 activation in CD8+ T cells suppresses IFNγ production, which, in turn, suppresses CXCL10 release from TAMs. STAT3 inhibition resulted in an increase in CXCR3-expressing CD8+ T cells and a better prognosis (70).
Trafficking of effector T cells and NK cells affected the release of pro-inflammatory cytokines (IFNγ, GMCSF, and TNFα) and chemokines (CCL4, CCL5, and CCL23) in the TME, assisting in the recruitment of extra effector immune cells and signaling of anti-tumorigenic pathways (71). For efficient tumor killing, the anti-tumor M1 macrophage expresses IL12, IL1, and iNOS (72). In several cancers, an elevated M1/M2 TAM ratio is associated with longer survival and better clinical outcomes, including small cell lung cancer (73), non-small cell lung cancer (74), colorectal cancer (75), ovarian cancer (72), breast cancer (76), oral squamous cell carcinoma (77), and others (78). LPS-induced TLR4 alone or in combination with IFNγ activates the PI3K-mTOR-AKT (phosphoinositide-3-kinase-mammalian target of rapamycin) pathway, which leads to the polarization of anti-tumor M1 macrophages and the suppression of cancer cell proliferation (79). Furthermore, phosphatase and tensin homolog (PTEN) regulates the inflammatory response via M1 polarization (80). NFκB (81) is a major regulator of M1 activation. TNFα is a positive regulator of M1 polarization and a negative regulator of M2 polarization when the NFκB pathway is activated. Furthermore, the myeloid differentiation primary response 88 (MyD88) suppresses M2 gene expression in TAMs, resulting in an anti-tumor M1 phenotype (82).
4.1 M1 macrophages in TMEM1-like macrophages perform their anti-cancer function by first efficiently distinguishing cancer cells from surrounding healthy cells by recognizing altered or cancer-specific antigens. Macrophages recognize altered carbohydrate structures (or glycosylation) that cancer cells occasionally present on their cell surfaces. Some tumor antigens, such as carcinoembryonic antigen and Tn antigen, are glycosylated molecules that lectin-like receptors on macrophage cell membranes recognize (83). Secondly, M1 eventually kills cancer cells via directly inducing cytotoxicity, phagocytosis, and antibody-dependent cell-mediated cytotoxicity (ADCC) (84).
The first step is somewhat slow, lasting 1 to 3 days and involving several stages such as the formation of ROS (reactive oxygen species) and RNS (reactive nitrogen species), as well as the production of IL1 and TNF to destroy cancer cells (85). Activated anti-tumor M1 macrophages target tumor cells by generating ROS and nitric oxide (NO), causing DNA damage, cytotoxicity, and apoptosis (86). M1 macrophages sustain themselves as well as induce NK cell and cytotoxic T-cell infiltration and activation in the tumor site by secreting substantial amounts of pro-inflammatory cytokines IFNγ and IL12 with anti-tumor activity, indicating an indirect mechanism of inhibiting cancer progression (87). Recognizing cancer cells and effectively phagocytosing them, on the other hand, suggests their ability as innate immune effectors, which later cross-primes adaptive immune response by presenting antigens on their surface (88). M1 uses ADCC as an adaptive response, employing anti-tumor antibodies for opsonization, which is a quicker process (89). After adhering to the Fc region of the antibodies, macrophages can phagocytose cancer cells coated with antibodies. To avoid M1-mediated elimination, tumor cells downregulate recognition molecules and activate counter-signals, such as PD1 activation in TAMs (88).
Anti-tumor M1 macrophages also stimulate the immune system’s anti-tumor activity, slowing cancer growth. Dectin1 expression on M1 macrophages increases NK cell-mediated tumor cell death (49). M1-like macrophages increase the number of both total and activated NK cells in the fibrotic liver, resulting in TRAIL-induced cell death (90). M1 also has increased co-stimulatory activity for effector T-cell activation (87). Other mechanisms include decreased VEGF, MMP, and CCL18 secretion relative to pro-tumor M2 macrophages, which prevents angiogenesis and metastasis (87). The key treatment options to target TAMs in the TME include TAM depletion, re-polarization into a more pro-inflammatory (M1) state, or reawakening of M1 macrophages’ anti-cancer potential (16).
4.2 TME shifts anti-tumor M1 to pro-tumor M2 phenotypeAs the tumor grows bigger, TME alters the ratio from M1 to M2 (71) (Figure 1). Malignant cells release M2-chemoattracting cytokines such as IL10, CCL2/CCL3/CCL4/CCL5/CCL7/CCL8, CXCL12, VEGF, platelet-derived growth factor (PDGF), and CSF1 to attract monocytes and M0 macrophages to the TME and differentiate them into M2 phenotype (72). TRMs are the first to be impacted by the pro-tumor TME machinery, resulting in an M2-rich tumor mass, which is frequently associated with a poor prognosis in numerous malignancies (91). Exosomes generated by HCC remodeled macrophages to the M2 subtype, causing pro-tumor responses in other immune compartments (92).
Interestingly, macrophage polarization in the TME is also correlated with distinct metabolic characteristics of glucose, lipid, and glutamine metabolism (93). Such metabolic alterations can determine the phenotype and functionality of TAMs in promoting cancer progression. Cancer cells utilize their metabolic by-products to alter the phenotype and functional activity of tumor-infiltrating immune cells for their growth. Lactic acid produced by the “Warburg effect” shifts the macrophages toward a more M2-like state (94). In ovarian cancer cell membranes, cholesterol efflux drives TAM reprogramming and tumor progression by inducing an IL4-mediated Th2-like environment (95). In addition, glutamate-ammonia ligase (GLUL) favors M2-like TAM polarization by catalyzing the conversion of glutamate into glutamine, whereas GLUL inhibition reverses the change (96). The recruitment and induction of macrophages in the TME can be significantly influenced by hypoxia. TAMs are drawn to and found in greater numbers in hypoxic areas of TME as a result of HIF1/2 and endothelin-2, released by hypoxic cancer cells (97). Damage-associated molecular pattern (DAMP), which is released as a consequence of hypoxia, induces the number of M2-TAM in the TME, which, in turn, secretes high levels of pro-angiogenic factor VEGF (98).
Other immune cells such as T-regulatory (Treg) cells, myeloid-derived suppressor cells (MDSCs), and B cells also control the polarization of macrophages. TAMs’ metabolic adaptability, mitochondrial integrity, and survival rate are indirectly, yet specifically maintained by Treg cells (99). In addition, MDSCs also regulate TAM differentiation in the TME (100). B cells can induce M2b macrophage polarization in human HCC, as well as suppress the function of M1 macrophages in the TME to promote the proliferation of cancer cells (101).
The polarization state of macrophages is a continuum and relates to the activation state of a macrophage at a specific moment, depending on the availability of numerous signals from other components of the TME (102). The M1 phenotype can transform into an M2 phenotype or vice versa in response to TME factors such as the accessibility of cytokines, growth factors, hypoxia, and the influence of other immune cells. There is more to macrophage divergence than the binary designation of M1 and M2, which represents the extremes on this spectrum and many intermediate cells may co-express both the markers of M1 and M2, indicating the complexity of polarization (103).
5 Pro-tumor M2-TAMs promote various aspects of tumor growth5.1 TAMs encourage increased tumor cell proliferationReplicative immortality being a crucial hallmark of cancer is enabled with increased infiltration of TAM in the case of many human malignancies such as breast cancer, endometrial cancer, and renal cell cancer (104). In vivo co-culture studies of macrophage and tumor cells highlighted the importance of infiltrated TAMs for the growth of tumor cells, and resultantly, depletion of TAMs effected the opposite (105).
Several growth factor receptors, such as EGFR and FGFR proteins, along with their associated signaling pathways, are frequently upregulated in a multitude of human malignancies, which ultimately result in heightened cellular proliferation and enhanced survival rates (106). TAMs employ this mechanism by producing growth factors like epidermal growth factor (EGF) and fibroblast growth factor (FGF) to facilitate the activation of downstream signaling pathways, subsequently leading to enhanced survival, migration, metastasis, and suppression of apoptosis in cancer cells (106) (Figure 2). TGFβ is widely recognized for its dichotomous function in the advancement of cancer, while serving as a suppressor of tumor growth during the initial phases and subsequently as a promoter of tumor development in later stages. Loss of ability in late-stage cancer to respond to cytostatic functions of TGFβ again results in increased cell proliferation, survival, angiogenesis, and immunosuppression (107). Stemming evidence indicates that TAM‐derived TGFβ promotes colorectal cancer progression through HIF1‐TRIB3 signaling (108). Additionally, TAMs secrete large amounts of immunosuppressive cytokine IL10, which prevents tumor cell-killing activity of CD8+ T cells, Th1 cells, and NK cells (109), thereby limiting cytotoxicity of the TME to help in tumor growth. Also, TAM-derived adrenomedullin interacts with endothelial cells to promote tumor growth through activation of the eNOS signaling pathway in a paracrine manner (110). Therefore, the development of tumors is critically related to the molecular cues shed by TAM.
Figure 2 M2-TAM promoting different aspects of tumor development: TAMs encourage increased tumor cell proliferation by secreting tumor cell proliferating growth factors such as EGF and FGF. TAMs cause neoangiogenesis and lymphangiogenesis by releasing various pro-angiogenic factors such as VEGF, PDGF, TGFβ, MMPs, and CXCL8. TAMs also lead to increased EMT and extracellular matrix remodeling by releasing factors such as CCL18, TGFβ, MMPs, and TNFα, which ultimately causes metastasis and secondary tumor formation. TAMs negatively affect the functions of NK cells, DCs, and cytotoxic T cells and promote immunosuppression by actively playing a role in the recruitment of Treg cells in the TME. Created with BioRender.com.
5.2 TAMs cause neoangiogenesis and lymphangiogenesisThe process of metastasis requires vascularization in primary tumors, which otherwise grow only up to 2–3 mm3 in size (106). TAMs encourage the sprouting of new blood vessels to provide oxygen and nutrients to proliferating cancer cells, described in animal models of ovarian cancer, cervical cancer, prostate cancer, breast cancer, and melanoma (111). Although tumor cells themselves provide the required stimuli to begin vascularization, the process is hindered in the absence of TAMs. Zeisberger et al. found that depletion of TAMs with clodronate encapsulated in liposomes (clodrolip) reduced blood vessel density in the tumor tissue (112). These results validate the idea that TAMs present in TME promote neoangiogenesis in tumors. Perpetuation of hypoxia and the formation of dysfunctional vessels are the by-products of tumor angiogenesis, as well (113).
TAMs are known to release a diverse array of molecules that are essential for the process of neoangiogenesis, which, in turn, plays a critical role in the onset of metastasis. For example, TAMs release growth factors such as VEGF, PDGF, TGFβ, and FGF, that promote angiogenesis (Figure 2) in numerous cancers, namely, gliomas, squamous cell carcinomas of the esophagus, breast, bladder, and prostate carcinomas (114, 115). Interestingly, angiogenic factors govern the process in a macrophage subtype context too; for instance, FGF signaling controls M2a-induced angiogenesis while placental growth factor (PlGF) signaling controls M2c-induced angiogenesis (55). Moreover, TAM-derived MMP1, MMP2, MMP3, MMP9, MMP12, plasmin, and urokinase plasminogen contribute to the cause of angiogenesis by degrading the ECM (116), which might later aid in metastasis. Accumulation of TAMs in tumor hypoxic regions and their adaptability to the low-oxygen microenvironment is particularly fascinating as they over-express pro-angiogenic factors (VEGF, pFGF, and CXCL8) and glycolytic enzymes, regulated by hypoxia-induced transcription factors HIF1 and HIF2 (109). In addition to this, HIF1-dependent chemokine CXCL12 is released by TAMs and serves as a potent chemoattractant to CXCR4-expressing endothelial cells, causing migration of endothelial cells, further helping neoangiogenesis (117). Furthermore, by closely collaborating with endothelial cells at the angiogenic branching points, M2-like macrophages aid the development of blood vessels by encouraging endothelial cells to condense into tubes, forming a tubular network (55). Cumulatively, TAMs and hypoxia both regulate one another in a positive feedback loop as hypoxia drives TAM polarization (97) and, on the other hand, TAMs favor hypoxia. TAM-led neoangiogenesis often amounts to abnormal blood vessel formation with irregular blood flow, laying the ground for hypoxia (118). In addition, such leaky blood vessels give way to tumor cell intravasation and metastasis (97). Interestingly, it has been recently reported that M2-derived exosomes transferring miR-193a-5p lead to tumor progression by endorsing vascular mimicry (119).
Similar to angiogenesis, lymphangiogenesis provides an additional avenue for malignant cells to travel through the body and establish footholds in other areas, hence predicting a poor clinical prognosis. TAMs promote lymphangiogenesis through the VEGFC/VEGFD-VEGFR3 signaling pathway (120) and frequently produce MMP9, leading to the development of lymphatic vessels. CD11b+ macrophages express lymphatic EGFs and induce lymphangiogenesis, either by trans-differentiating and directly incorporating into the endothelial layer or by stimulating the division of pre-existing local lymphatic endothelial cells (121). As a consequence, TAM-mediated vascular growth ensures survival of tumor cells at the local site along with paving the path for distant metastasis.
5.3 TAMs lead to epithelial–mesenchymal transition and extracellular matrix remodelingEpithelial–mesenchymal transition (EMT) is yet another cardinal aspect abetting metastasis wherein epithelial-like cancer cells lose the capacity for cell–cell adhesion and adopt a fibroblast-like phenotype with invasive and migratory properties, enabling cancer cells to leave the primary tissue site and enter the bloodstream or lymphatic vessels to infiltrate other organs (122) (Figure 2). Over two decades ago, Gorelik et al. conducted an in vivo study where they showed that TAMs play a significant role in tumor metastasis (123). Intravenous administration of murine tumor cells resulted in a notable augmentation in the TAM population at the site of lung tumor nodule development. Subsequent investigation has elucidated that EGF secreted by TAMs engages with CSF1 released by tumor cells facilitating the migratory capabilities of the tumor cells (124).
Hagemann et al. established that co-culturing TAMs with tumor cells promotes the expression of MMP2, MMP7, and MMP9 in a TNFα-dependent manner, causing the breakdown of ECM proteins and thereby helping metastasis (125). Furthermore, Seth et al. demonstrated that MMP7 could also encourage tumor metastasis through NFκB-RANKL signaling (126). MMP9 from M2-like TAMs causes collagen proteolytic clearance and lysosomal degradation. TAMs are also involved in the overexpression and reorganization of several collagen fibers, including collagen I and collagen VI, resulting in ECM deposition near aggressive tumor cells (127). Thus, TAMs remodel ECM surrounding cancer cells, which may protect them from external assaults while also degrading the ECM matrix to facilitate metastasis. TNFα and TGFβ produced by TAMs aid SNAIL and ZEB1 expression by activating the NFκB and β-catenin pathways respectively, adding to the EMT cause (128). Also, a positive feedback loop generated by tumor-shed GMCSF activates TAM to release CCL18, supporting EMT (129) (Figure 2). Fascinatingly, it has also been demonstrated that decreased expression of E-cadherin, a tumor-suppressor protein that acts antithetical to EMT and metastasis, is associated with increased expression of the M2 marker CD68 (130).
In breast carcinoma and lung adenocarcinoma models, TAMs are implicated in active ECM remodeling by working in close conjunction with CAFs to increase tumor cell intravasation (131). CAFs and TAMs have a mutually beneficial relationship that orchestrates tumor growth and progression (132). Prostate CAFs, for example, have been observed to be involved in monocyte recruitment and M2 phenotypic skewing. M2-like TAMs, on the other hand, trigger the mesenchymal transition of CAFs, hence increasing their pro-tumor activity. Cancer cells collectively influence the TAM–CAF interaction, resulting in increased metastasis and angiogenesis (133). When compared to normal breast-derived fibroblasts, invasive breast cancer-derived CAFs were able to drive monocytes towards the immunosuppressive M2 type. CAFs induced migration-related proteins in cancer cells while also influencing macrophage phenotype (134). The co-existence of CAFs with high numbers of TAMs was positively correlated in breast and oral cancers, serving as a marker for poor patient survival (132, 134). Similarly, CAF shed GMCSF and IL6, which both promote the pro-tumor TAM and cancer invasiveness (132). TAM-shed osteopontin also stimulates CAF to shed osteopontin to promote ECM modification and migration in HCC (135).
Regular tissue fibroblasts, secrete a diverse range of ECM ingredients such as collagen, elastin, fibronectin, and proteoglycans to build a network of fibers that acts as a barrier protecting tumor cells from mechanical, pharmacological, and immunological stress (132). CAFs, on the other hand, secrete fibroblast activation protein, a serine protease that remodels the ECM by rebuilding collagen and fibronectin into parallel orientation for improved pancreatic cancer cell motility and invasiveness (136). CAFs are also known to operate as leading cells that modify the ECM matrix and are closely followed by tumor cells that travel the paved way for them, causing metastasis (137). CAF expression of MMP11, MMP2, MMP9, and MMP21 in various malignancies is associated with a significant risk of tumor relapse (132). Furthermore, CAF-mediated ECM breakdown aids TAM infiltration into the tumor site. CAF-released ECM components likewise show an M2 bias (138, 139). TAM trafficking in TME is linked to the release of hyaluronan by stromal cells. Hyaluronan synthase-2 (HAS2) depletion in fibroblasts resulted in reduced macrophage recruitment, increased angiogenesis, and lymphangiogenesis at the tumor site (140). Thus, CAFs play an important role in regulating TAM polarization and mobilization, as well as encouraging ECM reorganization, which contributes to cancer migration.
Therefore, promising pieces of evidence describe TAM hallmarks and explore emerging mechanisms that contribute to their pathophysiological adaptations that promote tumorigenesis by fostering angiogenesis, invasion, and metastasis (141).
6 TAMs interact with other immune cells and cause immunosuppression in the TMEImmunosuppression in tumors makes it easier for cancer cells to evade immune surveillance (142). Studies have linked the role of different cytokines and molecules secreted by TAMs, such as TGFβ, IL10, and arginase-1, along with the activation of certain signaling pathways to play a significant role in immunosuppression (7). TGFβ influences macrophage polarization in TME during the innate immune response, favoring the M2 phenotype, which again encourages the synthesis of additional TGFβ (143). TGFβ also prevents the cytolytic activity of NK cells (7), while both TAM-derived IL10 and TGFβ inhibit the maturation and functioning of dendritic cells (DCs), along with inducing apoptosis. This results in reduced antigen presentation and dampened adaptive immune response (144). TGFβ regulates the production of various cytolytic genes as well, including granzyme A, granzyme B, IFNγ, and FAS ligand, thereby impairing the anti-tumor activity of CD8+ T cells during the adaptive immune response and ultimately resulting in pro-tumorigenic TME (7). Moreover, TAMs can directly promote cytotoxic T-cell exhaustion. Infiltrating CD8+ T cells are observed to have greater effector potential after in vivo TAM reduction (145). TGFβ is also associated with supporting the differentiation of Treg cells, a powerful immunosuppressing arm of the TME (146). TAM-mediated Treg recruitment has been reported in ovarian cancer, nasopharyngeal carcinoma, and liver cancer. Such recruited Tregs deactivate cytotoxic T cells against cancer (145). On the other hand, TAM-derived IL10 suppresses the expression of anti-tumor cytokines IL12 and IFNγ, involved in naïve T-cell differentiation, and further inhibits the function of cytotoxic T cell, NK, and lymphokine-activated killer cells (Figure 2) (145). It has been shown that arginase-1, a marker for M2 macrophages, converts L-arginine into polyamine and proline, causing the TCR signal to be dysregulated, and resultantly causing CD8+ T-cell anergy (147). Additionally, cues provided by TAMs lead to the expansion of GR1+CD11b+MDSCs, which overexpresses iNOS, arginase, and TGFβ, directly contributing to immunosuppression (148). Multiple TAM-produced chemokines have been related to immunosuppression as well. For example, TAM-derived CCL17/CCL22 significantly aids in infiltration of Tregs to the TME through the chemokine receptor CCR4 (149). CCL18 produced by TAM attracts naïve T cells to the tumor site, resulting in T-cell anergy (114).
Another effective mechanism M2-TAMs employ to deplete the anti-tumoral immune response and carry forward its immunosuppressive streak is by displaying an amplified expression of immune checkpoint molecules PDL1 and cytotoxic T-lymphocyte antigen 4 (CTLA4) ligand (150). Heightened levels of these molecules are correlated with poor prognosis in various cancers such as HCC and glioblastoma (151); therefore, they present themselves as secondary targets for current immunotherapies, by using ICI such as pembrolizumab or nivolumab (152). This discussion clarifies how M2-TAM promotes different aspects of tumor development (Figure 2). Interestingly, inflammation in the TME is also associated with cancer progression. TAMs and monocytes contribute to this inflammatory process by the generation of IL21+ T follicular helper cells (TFH cells). This creates a local environment suitable for the differentiation of plasma cells and M2b macrophages, which promotes cancer progression (153). TAMs also establish an inflammatory milieu via releasing numerous pro-inflammatory cytokines such as CXCL8 in endometrial cancer, IL6 in breast cancer, and IL1β in pancreatic cancer, which ultimately have correlated with poor prognosis (145).
However, focusing on TAMs and their interaction with only cancer cells and immune components is addressing half of the story. The other half is the interplay of CSC–TAM, which may be the cardinal cause of tumor progression and survival, owing to the tumor-initiating, metastatic, and relapse properties of CSCs and the TAMs that contribute to the major population of TME and their pro-cancerous inclination.
7 Cancer stem cells and TAMsAs previously pointed out, the rare subpopulations of CSCs are the drivers of an entire tumor mass due to their tumor-initiating properties. According to the CSC theory, CSC resides on top of the hierarchal model in tumor development wherein, much like somatic stem cells, they undergo asymmetrical division to generate differentiated NSCCs sculpting the tumor, as well as self-renew to maintain itself (154, 155). CSCs not only develop a tumor at its primary site, but they also metastasize to the secondary site to create a new tumor (4, 156). All disseminated cells are not able to make it through the metastatic journey or can successfully break away from ECM from the primary site (157, 158). The exceptional properties of CSCs to modulate ECM and undergo EMT, and their inherent quiescent nature, make them sturdy for this tedious journey (157, 159). A previous report from our group has elucidated how intrinsic non-migratory CSCs give rise to metastatic CXCR4+ CSCs, which indeed are responsible for the seeding of new tumors at secondary sites (160). Additionally, resistance to chemo- or radiotherapy and causing further relapse to the same or distant sites are attributed to CSCs as well (161, 162). CSCs, but not NSCCs, successfully escape these treatment regimens owing to their self-protecting non-proliferating quiescent state and overexpression of drug resistance pumps (163, 164). Intriguingly, chemotherapy leads to an increase in this CSC subpopulation which may later “recreate” more aggressive tumors after therapy removal, explaining the failure of eradicating cancer in many cases (165). However, the pivotal role that CSC plays especially in tumor initiation, metastasis, and tumor recurrence would not have been possible, if it did not evade the anti-tumor immune response first and further carry out immune-editing in its favor (166).
CSCs hamper the anti-immune components of their niche to ensure their existence. For example, CSCs inhibit anti-tumor NK cells by inducing apoptosis on one hand, while moderating their cell surface receptors to avoid detection on the other (23, 167). Similarly, CSCs avoid CD8+ cytotoxic T-cell immune surveillance by downregulating MHC-I expression and in turn, impede their growth by selectively augmenting the inhibitory checkpoint receptors (23, 168). Additionally, CSCs promote a pro-tumor environment by recruiting immunosuppressive MDSCs and T-regulatory cells, known to release cytokines and bring about metabolic changes that not only help in CSC maintenance but also empower their selective survival over the others (14, 169, 170). The existence and the position of TAMs are observed to be positively correlated with that of CSCs. Numerous reports suggested that the distribution of infiltrating TAMs in numbers and being in the vicinity of CSCs are also directly associated with histological grade and the number of CSCs present (7, 171). Similarly, TAM accumulation was also clearly evident within the hypoxic regions of tumors (13), which correlates with the existence of CSCs as well in these hypoxic regions (172). Further studies suggested that CSCs and TAMs, when introduced simultaneously in wild-type syngeneic mice models, depict more efficient tumor initiation and metastatic activities (173). Supporting the positive reciprocal cross-regulation of CSCs and TAMs, another finding showed heightened tumor formation capability in xenograft models when TAMs were co-cultured with hepatocellular CSCs first (2, 174). Considering that TAMs constitute the dominant proportion of the tumor milieu and their close partnership with CSCs, their dialog in TME needs to be understood further.
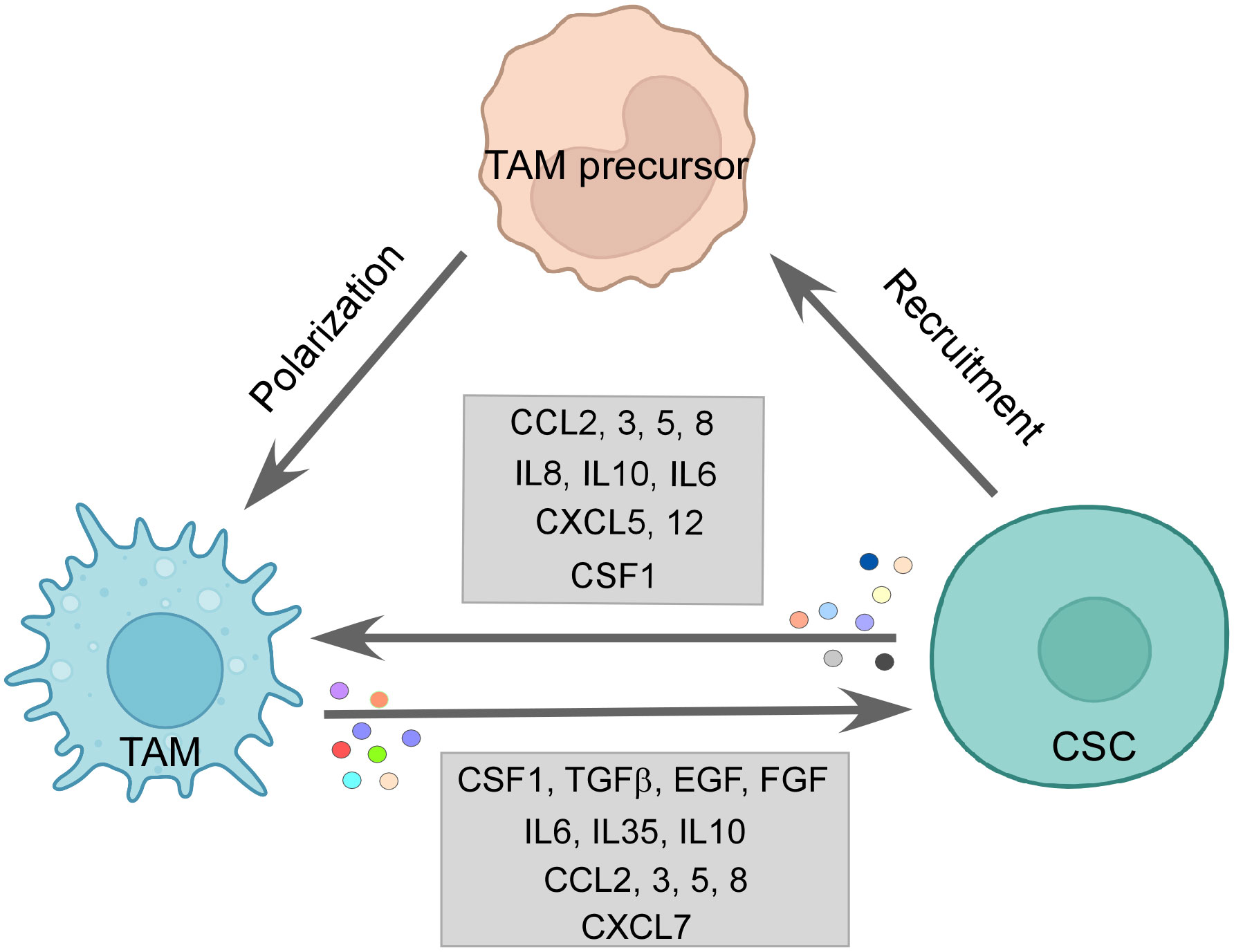
7.1 CSC effect on TAMsCSCs release chemotactic factors such as CCL2, CCL3, CCL5, CCL8, CXCL5, and CXCL12 dynamically to recruit macrophages or their precursors (7, 175). It has been observed in a multitude of cancers that CCL2 is expressed in higher amounts by CSCs than non-CSC counterparts. Along with this, higher expression of CCL2 correlated with higher engagement of TAMs, especially the M2 subtype to the tumor site, while blocking of the same impaired the M2 population recruitment (175, 176). Interestingly, the production of CCL2 is influenced by the protein TWIST, which is known to be upregulated in CSCs (177). CCL5 is seen to be exclusively secreted by CSCs (175). Although photodynamic therapy (PDT) elicited CCL8, which actively brought about an M1 anti-tumor response in cutaneous squamous cell carcinoma (178), CCL8 is also reported to enhance stemness in cancers (179). Both CXCL5 and CXCL12 are expressed by CSCs, which readily encourages TAM infiltration. CXCL5 being downstream of Sox9 attracts both neutrophils and macrophages, while CXCL12 activates CXCR7, which regulates metastasis in breast cancer as well as M2 infiltration (175). Interleukins such as IL33 are also secreted by CSCs to create a pro-tumorigenic environment by attracting TAMs to their site (180). Some soluble factors, such as CSF1 and VEGFA, are also reported to be involved in TAM recruitment. Glioma CSCs are reported to release CSF1 and cytokines not only to recruit macrophages but also to polarize them to an immunosuppressive type (7, 175) (Figure 3).
Figure 3 The CSC–TAM crosstalk: CSCs employ various modalities to recruit M2-TAMs, such as CCL2, CCL5, CCL8, and CSF1. M2-TAMs are also polarized in the tumor site by CCL2, CCL5, IL6, IL10, and CSF1 by CSCs. Reciprocally, TAMs promote CSC function by releasing CSF1, TGFβ, IL6, IL10, IL35, EGF, and FGF among others, thereby, maintaining a vicious loop for sustenance of one another. Created with BioRender.com.
Similarly, CCL2 and CCL5 released by CSCs contribute to both macrophage infiltration and skew them towards the tumor-supporting M2 subtype (181, 182) (Figure 3). A multitude of cytokines participate in this process of altering the functional state of the macrophages in favor of CSCs; for example, IL6, IL8, and IL10 released by the CSCs are all reported to employ STAT3 signaling to convert TAMs to M2 phenotype (175). IL10 being highly expressed in CSCs is also known to engage NFκB to polarize M2-like macrophage (183). Moreover, IL4 and IL13 released by the CSCs also bring about the pro-tumor state of macrophages (12). Again, CSF, a recruiter for macrophages, also supports the M2 phenotype along with the CSC-released immunosuppressive cytokine TGFβ (184). CSC-derived exosomes are reported to carry out the same effect as well. For instance, glioblastoma stem cell-derived exosomes skewed monocytes to pro-tumor M2 macrophage by transferring them with STAT3, the activation of which we have already seen to induce the pro-tumor TAMs (185). Moreover, CSCs can push macrophages toward M2 by direct contact-dependent mechanisms as well (175), therefore elucidating the varied ways CSCs use to exercise their immunomodulatory arm.
Apart from manipulating immune cells on its side, CSCs also evade immune-mediated destruction. CSCs express low levels of MHC class molecules and co-stimulatory receptors, which may otherwise trigger immune responses (186). Additionally, immune checkpoint ligands PDL1 and B7-H3 are overexpressed on CSCs to limit the growth of immune cells (12). Another notable immune evasion mechanism employed by CSC, especially against phagocytosis, is the overexpression of “don’t eat me marker” CD47, the interaction of which with SIRPα on M1 macrophages stills the phagocytosis process (12, 13). Furthermore, it has been observed that M2-like TAM expresses SIRPα at a noticeably higher level and interacts with the upregulated CD47 in CSCs (187, 188).
7.2 TAM’s effect on CSCsReciprocally, the M2-TAMs help in CSC maintenance, growth, and EMT as well. Incidentally, the chemokines used by the CSCs to promote TAMs are also secreted by TAMs to help CSCs (12). CCL2 via the β-catenin pathway upregulated CSC-related stemness genes along with increasing ALDH and CD44 expressing CSC pool in breast cancer (181). Also, CCL3 released by TAMs induced migration and invasion characteristics (189). Similarly, CCL5 and CCL8 secretion by TAMs promoted stemness and invasion features in glioblastoma (179, 190). CXCL7 releasing TAMs, recruited by glioma, helps in regulating the stemness in such cancer cells (191). In HCC and breast cancer, IL6 from TAMs activates the STAT3 pathway to promote CSC population, migration, and angiogenesis (174, 192). TAMs also shed IL10 and IL35, which endorses the CSC nature. For example, IL10 from M2-TAMs induces the upregulation of stemness genes in lung cancer. Inhibition of the same presented as a potential therapeutic target (175). Therefore, observably IL6 and IL10 act in a pro-tumor immunosuppressive loop in TME where both M2 and CSCs secrete them, supporting the growth of one another. Interestingly, not only M2 but M1-TAMs are also reported to secrete IL6 and IL12, which nudge the tumor cells towards CSC phenotype (175). Moreover, factors like TGFβ, EGF, and FGF secreted by TAMs also skew cancer cells to CSCs. Fascinatingly, CSF1 and EGF establish a feedback paracrine loop between CSCs and TAMs, where CSF1 recruited TAMs shed a higher amount of EGF leading to stimulation of STAT3/SOX2 pathway in breast cancer cells (12, 193) (Figure 3). Talking of feedback mechanisms, CSC encourages TAM to release MGF-E8, which, via STAT3 and sonic hedgehog pathways, induces CSC-led tumor formation and resistance to chemotherapy drugs (194). M2-derived extracellular vesicles deliver miR-21-5p, which maintains CSC stemness and characteristics in pancreatic cancer (195).
TAMs impact CSC growth by contact-dependent mechanisms as well. Metastatic CSCs express HAS2, which facilitates the interaction between CSCs and TAMs, leading to secretion of PDGF-BB by TAMs. This factor, in turn, orchestrates the secretion of FGF7 and FGF9 from bone stromal cells, enabling CSC growth (196). The physical interaction of CD90 and Ephrin type A expressed on CSCs with their respective ligands on macrophages activates signaling pathways, thereby enhancing high tumorigenicity and metastasis by CSCs (173). Another juxtacrine interaction of LSECtin on TAMs and BTN3A3 receptor on CSCs drives tumor stemness (197). GPNMB on TAMs binds to the CD44 of tumor stem cells, eliciting a response of IL33, which is a proponent of CSC growth. In addition, IL33 instigates macrophages to release pro-CSC cytokine TGFβ (12). Consequently, the dynamic feed-forward equation between CSCs and TAMs drives the population of each other and creates an immunosuppressive environment, which consequently supports the survival of both populations (Figure 3).
8 Therapeutic interventionConsidering the crucial role played by TAMs in promoting pro-cancerous TME, targeting these TAMs becomes imperative for alleviating their tumor-reinforcing arm for better anti-cancer therapy outcomes. This can be achieved by depletion of TAM, blocking TAM infiltration, and polarizing TAMs to M1 phenotype, which resultantly leads to activation of anti-tumor activity, overcoming TAM-mediated immunosuppressive TME and engaging their phagocytosis potential (2, 11).
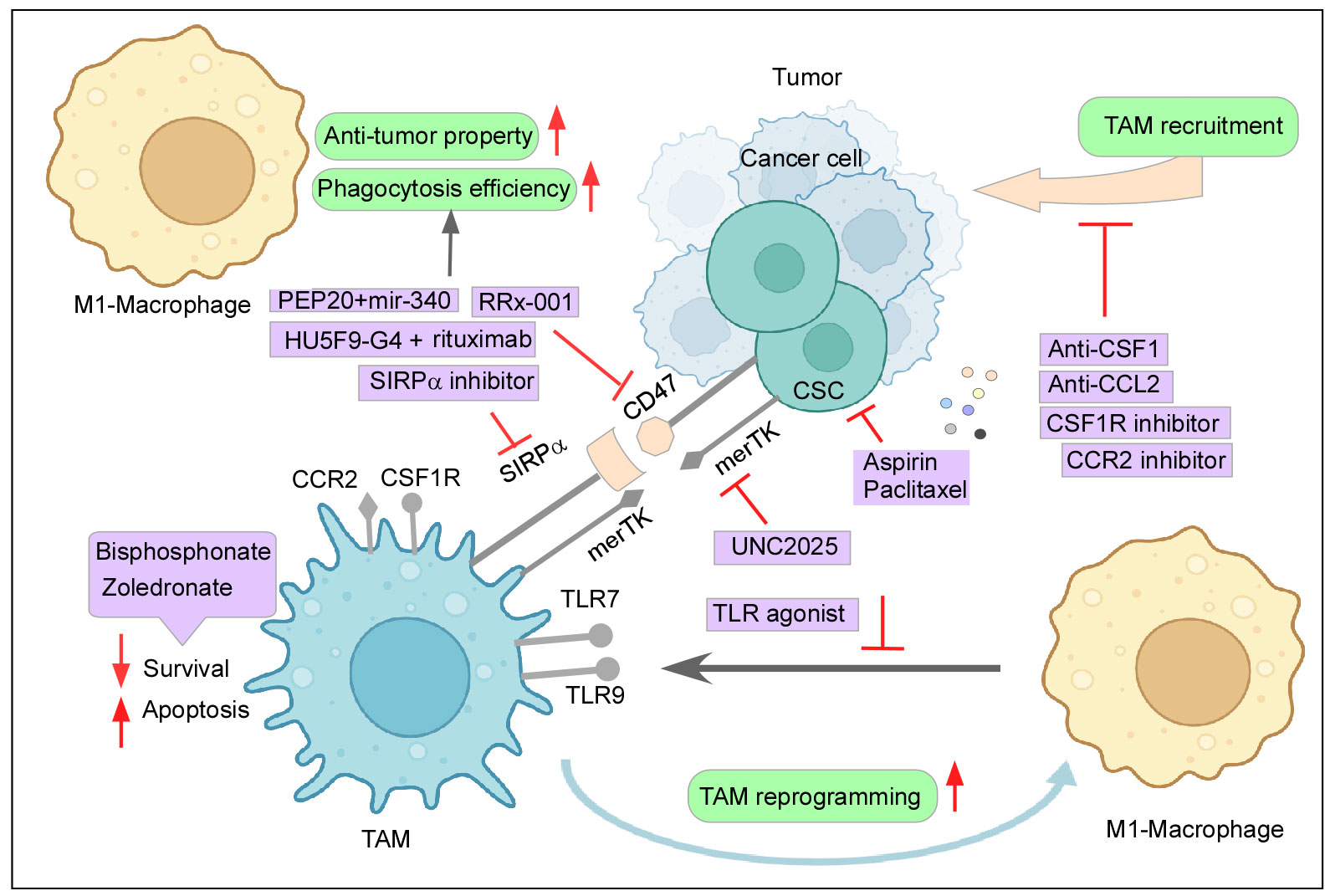
8.1 Depletion and inhibiting infiltration of TAMs in TMEBisphosphonates are known to induce TAM apoptosis and suppress the migration and infiltration of TAMs (11). Zoledronate, a class of bisphosphonate, can be specifically delivered to attack TAMs by nanoparticle systems and has shown promising results of reduced immunosuppressive effect and tumor growth in mice models (11, 198). Chronic stress-induced macrophage infiltration and higher expression of PDGF-AA in orthotopic ovarian cancer models create an inflammatory environment with negative implications. This stress-induced unfavorable effect can be reversed by Zoledronic acid, a macrophage-depleting drug (199). Furthermore, macrophages generated from the peritoneal cavity of mice with subcutaneous pancreatic cancer promoted tumor growth and metastasis, which Zoledronic acid successfully inhibited (200). Zoledronic acid coupled with doxorubicin was effective against breast and peritoneal TAMs in both in vitro and in animal models (201) (Figure 4). As seen earlier, CSF plays a paramount role in TAM recruitment and survival, and therefore, blockading of the CSF1/CSF1R axis can reduce monocyte recruitment to the tumor site and its differentiation while further hampering the survivability of the existing TAMs (6, 11). Interestingly, monoclonal antibodies and small-molecule inhibitors targeting CSF1R have been reported to result in a decreased number of TAMs and an increased ratio of CD8+/CD4+ T cells in the tumor (9, 202). TD-92, an erlotinib derivative targeting CSF1R, not only depletes TAM at the tumor site but also increases the efficacy of anti-PD1 therapy (203). Monoclonal antibodies against CSF1, Lacnotuzumab (NCT02435680), with chemotherapy showed improved patient responses compared to chemotherapy alone in advanced triple-negative breast cancer in a recent randomized phase II clinical trial (204). Similarly, LY3022855 (NCT01346358) showed decreased TAM levels in solid refractory tumors (205) (Figure 4). Despite the few upsides reported by this therapy, numerous accounts state otherwise. Long-term blockade of CSF1R led to PI3K activation, which led to therapy failure (206). Impairing CSF1R axis may also have a limited durable response due to infiltration of Tregs in the tumor milieu and inhibition of TRMs, which are crucial elements for a body’s immune equilibrium. Moreover, in contrast to some previous studies, attenuating CSF1R along with PD1 immunotherapy failed to produce significant positive outcomes in patient clinical trials (6, 207).
Figure 4 Therapeutic intervention to curb the tumor–TAM interplay: Mechanisms to inhibit the cancer-TAM crosstalk include depletion and blocking M2-TAM recruitment, re-programming of M2-TAMs, engaging phagocytic activity, and dysregulating CSC–TAM crosstalk. Created with BioRender.com.
Similarly, the CCR2–CCL2 axis also provides itself as an attractive therapeutic strategy due to its reported role in active monocyte recruitment. Aiming this signaling pathway has been shown to reduce tumor growth, decrease the incidence of M2-TAM at the primary site and metastatic sites as well, and elicit an anti-tumor T-cell response (11, 208). In glioblastoma animal models, CCR2–CCL2 inhibitors effectively reduced the M2-TAM infiltration at the tumor site, improved survival, and demonstrated a better outcome of combinatorial anti-cancer therapies (11, 209). A clinical trial (NCT01413022) involving CCR2 inhibitor PF-04136309 in association with chemotherapy was observed to be safe and tolerable for locally advanced pancreatic cancer (210). When compared to chemotherapy alone, a CCR2 antagonist, CCX827 (NCT02345408), coupled with a chemotherapeutic drugs, FOLFIRINOX, improved patients’ overall survival in advanced pancreatic cancer (211). Similarly, Carlumab (NCT01204996), a monoclonal antibody against CCL2, displayed transitory effectiveness against tumor (212) (Figure 4). However, the withdrawal of the anti-CCL2 treatment regime exacerbated monocyte infiltration and metastasis (213). In addition to this, blocking of this axis did not affect the already recruited TAMs as they continued to aid tumor progression (18). Abating other monocyte recruitment strategies has delivered some positive results. Restricting the CCL5/CCR5 axis-mounted anti-tumor response lowers TAM trafficking and reduces tumor growth (214). Similarly, attacking IL8/CXCR2 generated higher efficacy towards PD1 treatment (215). Targeting the CXCL12–CXCR4 pathway along with chemotherapy also provided some relief in tumor burden by reducing M2-TAMs and tumor revascularization (216). Recent clinical trials employing CXCR4 antagonist LY2510924 in combination with durvalumab have shown a safe and manageable response in a phase 1 clinical trial (NCT02737072) with advanced refractory solid tumor (217). Furthermore, the CXCR4 inhibitor motixafortide in combination with pembrolizumab (anti-PD1 antibody) and chemotherapy demonstrated excellent benefits in patients with advanced pancreatic cancer (COMBAT/KEYNOTE-202 study NCT02826486) (218).
8.2 Reprogramming TAMs to anti-tumor M1 phenotypeReprogramming M2-TAMs to pro-inflammatory anti-tumor M1 subtype also presented itself as an attractive targeting strategy in this regard. Inhibiting CSF1R not only blocks TAM recruitment but also skews M2 to M1 phenotype, which effectively resulted in retarded tumor growth, inducing cytotoxic T-c
留言 (0)