
記住我

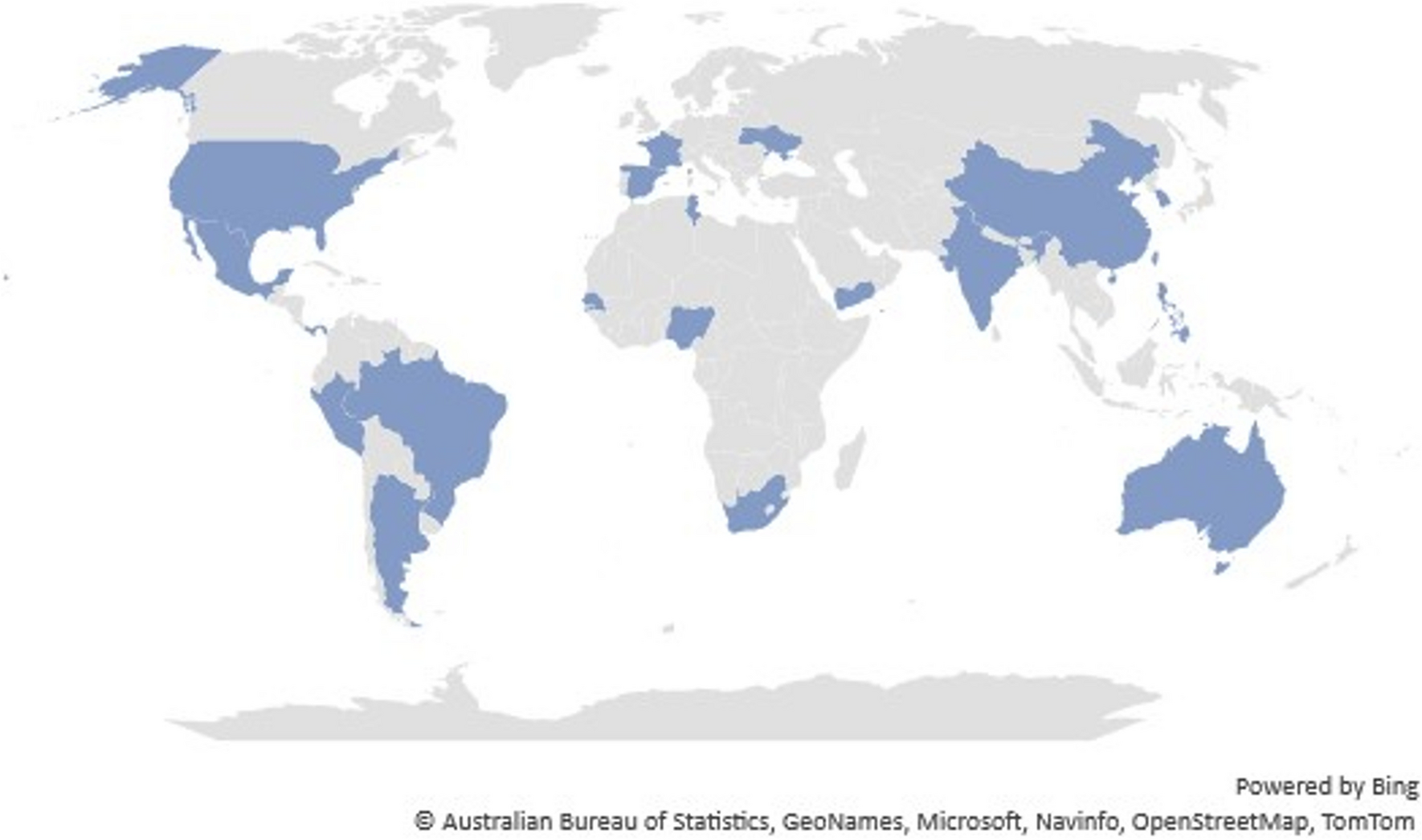

HSV-1 infection of oral, ocular, or nasal cavities leads to life-long latent infections in neurons within trigeminal ganglia (TG), brainstem, and other parts of the CNS, reviewed in [1, 2]. The latency-reactivation cycle is customarily divided into 3 stages: establishment, maintenance, and reactivation. The hallmark of establishing and maintaining latency is lytic cycle viral gene expression is silenced, infectious virus is undetectable, and neurons survive infection. During maintenance of latency, viral DNA is organized as chromatin, which does not support high levels of lytic cycle viral gene expression [3], and the viral genome is circularized. In contrast to productive infection, the latency-associated transcript (LAT) is the only viral transcript abundantly expressed during latency. LAT is a complex locus that expresses several micro-RNAs, a stable long non-coding RNA, and 2 novel small non-coding RNAs, reviewed in [1, 2]. LAT gene products inhibit apoptosis [4,5,6,7,8] and expression of viral genes important for productive infection [8,9,10]. Hence, LAT promotes neuronal survival and sustains a pool of latently infected neurons that can reactivate from latency multiple times in a mouse model of infection [11].
Approximately 400,000 individuals in the USA suffer from HSV-1 ocular disease. Recurrent eye disease, for example, herpetic stromal keratitis [12, 13], causes tissue destruction and occasionally blindness [14]. Acyclovir treatment only reduces recurrent ocular disease by 41% [14] because most cases are due to reactivation from latency [15]. HSV-induced encephalitis (HSE) is the most common cause of sporadic and fatal encephalitis [16, 17]. Although HSE generally occurs in the temporal and frontal lobes, HSE can also occur in the brainstem [18, 19]. The majority of HSE cases are due to reactivation from latency [16, 17].
Stressful Stimuli Correlate with Increased Episodes of Reactivation from LatencyStress (acute, episodic acute, or chronic), fever, UV light, and heat stress increase the incidence of reactivation from latency in humans [20,21,22]. Surprisingly, these divergent stimuli activate the glucocorticoid receptor (GR). For example, stress increases cortisol, which activates GR via a liganded mechanism [23]. Furthermore, an inhibitor of cortisol production impairs heat-shock induced HSV-1 reactivation suggesting heat stress increases cortisol levels [24]. Thirdly, UV light induces GR phosphorylation and transcriptional activation via ligand-independent mechanisms [25, 26]. UVB and UVC light, but not UVA, increase cortisol production in human skin cultures, and UVB light increases corticosteroid production in C57BL/6 J mice [27, 28]. Finally, UV light triggers expression of certain enzymes regulated by GR activation. In summary, these different reactivation stressors share common signaling proteins, including GR activation.
The stress response is primarily mediated by secretion of glucocorticoids, including cortisol, via the hypothalamic-pituitary adrenocortical (HPA) axis, reviewed in [23]. Cortisol diffuses across the plasma membrane and interacts with GR. The GR-hormone complex disengages from the heat shock protein (HSP) complex, and the GR-hormone complex enters the nucleus. A GR-hormone homodimer binds to a consensus GR response element (GRE), remodels chromatin, and stimulates transcription, [29, 30] (Fig. 1A; ligand-dependent activation). This process occurs within minutes and does not require de novo protein synthesis. A GR monomer can also stimulate transcription by binding certain 1/2 GREs [31, 32]. Notably, GR can also stimulate gene expression via an un-liganded mechanism [25] (Fig. 1B). For example, GR phosphorylation at serine 134 is important for ligand-independent GR activation, culminating in gene expression (25). Serine 134 is hyperphosphorylated following glucose starvation, oxidative stress, UV irradiation, and osmotic shock suggesting cellular stressors directly induce GR phosphorylation at serine 134. The GR can be phosphorylated by mitogen-associated protein kinases (MAPKs), cyclin-dependent kinases (CDKs), glycogen synthase kinase 3 beta (GSK3B), and likely additional protein kinases [25]. Although the mineralocorticoid receptor (MR) can also bind cortisol, we predict MR is not as important during reactivation because MR does not activate transcription as efficiently as GR [33]. Approximately 50% of TG sensory neurons express GR [34] suggesting GR activation has the potential to directly induce reactivation from latency by stimulating viral gene expression. For example, GR can function as a pioneer transcription factor in vivo by interacting with nucleosomal sites and recruiting Brg1, which culminates in remodeling nucleosomes [35]. The hallmark of a pioneer transcription factor is they bind silent chromatin, activate transcription, and cell reprogramming [36, 37].
Fig. 1
Activation Of Gr By Corticosteroids And Protein Kinases. A Schematic of key events that lead to GR activation by increased glucocorticoids secreted via the HPA. Red nucleotides in the GRE are essential nucleotides, capital letters are well conserved nucleotides, small letters are flexible, and N can be any nucleotide. A GR-hormone dimer specifically binds to a consensus GRE. A GR-hormone homodimer can also bind to a 1/2 GRE and stimulate transcription. B Certain protein kinases described in the text can phosphorylate GR, which promotes release of GR from the HSP complex (phosphorylated GR is denoted as GR-P). A phosphorylated GR dimer or GR monomer enters the nucleus, binds a consensus GRE or 1/2 GRE respectively, and transactivates promoters containing a 1/2 GRE. BioRender was used to generate this figure
Corticosteroids also have anti-inflammatory and immune-suppressive effects, in part because GR binds two transcription factors: activator protein 1 (AP-1) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-KB), reviewed in [38, 39]. The AP-1 transcription factor can be a homodimer or a heterodimer and is comprised of four sub-families of transcription factors: Jun (c-Jun, JunB, JunD), Fos (c-Fos, FosB, Fra1, Fra2), Maf (musculoaponeurotic fibrosarcoma; c-Maf, MafB, MafA, Mafg/f/k, Nrl), and ATF (activating transcription factors; ATF2, LRF1/ATF3, BATF, JDP1, JDP2) protein families. AP-1 transcription factors bind a consensus DNA sequence (TGA(G/C)TCA), and the most common heterodimer bound to the consensus site is c-Fos and c-Jun. AP-1 regulates numerous immune checkpoints, including T cell activation, expansion of T helper subsets, and co-stimulation of T-cell responses, reviewed in [40]. GR-mediated inhibition of AP-1 transcriptional activity occurs, in part because GR directly interacts with the c-Jun subunit of Ap-1 [38, 39].
The NF-κB/Rel family includes NF-κB1 (p50/p105), NF-κB2 (p52/p100), p65 (RelA), RelB, and c-Rel, reviewed in [39, 41, 42]. Most members of this family can homodimerize or form heterodimers with each other. The most common activated form of NF-κB is a heterodimer consisting of p50 or p52 subunit and p65. NF-κB is generally localized in the cytoplasm and is inactive because it is associated with regulatory proteins called inhibitors of κB (IκB). In the canonical pathway, tumor necrosis factor-α for example, IκB is phosphorylated and subsequently degraded: consequently, NF-κB rapidly enters the nucleus, binds to promoters containing the consensus binding site 5′-GGGRNYY YCC-3′ (R is a purine, Y is a pyrimidine, and N is any nucleotide), and activates expression of numerous genes that encode innate immune or pro-inflammatory regulators. GR inhibits NF-KB-dependent transcription by directly interacting with p60, recruiting histone deacetylases to NF-KB-dependent promoters, and/or preventing phosphorylation of the C-terminus of RNA Pol II [38, 39]. Finally, corticosteroids can readily induce apoptosis in certain lymphocyte subsets, which will reduce immune responses [38, 39]. In summary, increased corticosteroid levels are predicted to increase the incidence of reactivation by more than one mechanism.
Viral Proteins Predicted to Mediate Early Stages of Reactivation from LatencyWhen TG cultures obtained from mice latently infected with HSV-1 are infected with an adenovirus vector that expresses ICP0, ICP4, or virion protein 16 (VP16), reactivation from latency is induced [43]. These viral proteins are key viral transcriptional regulators and possess functions required to initiate lytic cycle viral gene expression during reactivation from latency. For example, ICP0 is a relatively large protein,110 kDa, that stimulates immediate early (IE), early (E), and late (L) HSV-1 gene expression (44); disrupts nuclear domain 10 structures [45]; and evades host intrinsic and innate antiviral defenses [46, 47]. Furthermore, ICP0 possesses E3 ubiquitin ligase activity that is crucial for its functions, reviewed in [48]. The ICP4 protein, a 175 kDa phosphoprotein, specifically binds multiple sites on the viral genome [49] where it recruits the TATA box-binding protein and RNA pol II transcription factor IIB to activate early and late viral gene expression (50). Consequently, ICP4 is essential for productive infection [51], and its expression triggers production of infectious virus during reactivation from latency. ICP0 and ICP4 mRNA are expressed as IE genes during productive infection; hence, these viral proteins are readily detected early after infection. The viral tegument protein (VP16) is expressed as a leaky-late protein that interacts with two cellular proteins: host cellular factor 1 (HCF-1) and Oct-1. This multi-protein complex binds specific sequences in IE promoters and transactivates all IE promoters, reviewed in [52,53,54]. Thus, expression of ICP0, ICP4, or VP16 could initiate lytic cycle viral gene expression and virus production during reactivation from latency.
HSV-1 Models for Studying the Latency-Reactivation CycleAn in vivo heat-stress model of reactivation from latency in TG neurons of mice concluded VP16 is essential for reactivation [55, 56]. A rat primary superior sympathetic neuronal model of latency also concluded VP16 initially drives reactivation from latency [57, 58]. This model predicts two phases drive production of infectious viruses [58]. The hallmark of the first phase includes de-repression of silent lytic viral genes, and this phase does not require viral proteins. The hallmarks of the second phase include nuclear localization of VP16 and host cell factor 1 (HCF-1), an essential VP16 transcriptional coactivator. These two phases precede increased viral gene expression and production of infectious virus. This same rat model reported ICP0 expression occurs after VP16 because ICP0 expression overcomes interferon treatment [59]. Additional rodent neuronal cell models of latency have provided insight into certain aspects of the HSV-1 latency-reactivation cycle, reviewed in [60]. Human embryonic neuronal precursor cells, Lund human mesencephalic (LUHMES), proliferate when expression of a tetracycline-regulatable (Tet-off) v-myc transgene is induced, and these cells can support HSV-1 latency [61]. LUHMES can be readily differentiated into neuronal-like cells, and the virulent HSV-1 strain (17syn +) reactivates more efficiently than a less virulent strain (KOS) [62] indicating cell culture model of latency is a useful model to compare to results from rodent neuronal models. Many of the cell culture models of latency require treatment of the antiviral drug, acyclovir, to stop viral gene expression and establish a quiescent infection.
When TG from mice latently infected with HSV-1 are dissected, minced into smaller pieces, and then placed in media, virus shedding consistently occurs, and this procedure is referred to as explant-induced reactivation. During explant-induced reactivation, LAT gene products are reduced [63], HCF-1 is rapidly recruited to IE promoters [64], chromatin remodeling of the ICP0 promoter occurs, ICP0 transcription occurs [65], and infectious virus is produced. The synthetic corticosteroid dexamethasone (DEX) accelerates explant-induced reactivation [66, 67], and a GR-specific antagonist, CORT-108297, impairs reactivation [67] indicating GR activation is important for this process. Immunohistochemistry studies revealed VP16 is detected prior to ICP0 and ICP4 during DEX-induced reactivation from latency [67]. Assuming these antibodies are equally effective for detecting viral proteins in formalin-fixed and paraffin-embedded thin sections, this finding appears to support the concept that VP16 is expressed early during reactivation from latency. An independent study using TG explants concluded that viral RNA expression is disordered during explant-induced reactivation [63].
Rabbits latently infected with the McKrae strain, a neurovirulent HSV-1 strain, undergo spontaneous reactivation from latency [68] or reactivation induced by iontophoresis [69]. UV light also triggers reactivation from latency in mice latently infected with HSV-1 [70]. Notably, DEX treatment of calves or rabbits latently infected with bovine alphaherpesvirus 1 (BoHV-1) is the only α-herpesvirus member where reactivation from latency is reproducibly initiated [71, 72]. Cell culture models of latency and explant-induced reactivation are important: however, they may not recapitulate all complex virus-host interactions that occur during in vivo reactivation.
Identification of Stress-Induced Cellular Transcription FactorsUsing transcriptomic approaches, stress-induced cellular transcription factors were identified in TG when calves latently infected with BoHV-1 are treated with DEX, which consistently initiates rapid reactivation from latency [73]. Expression of Krüppel like factor 4 (KLF4), KLF6, KLF15, promyelocytic leukemia zinc finger (PLZF), Slug (also referred to as Snail homolog 2), and Sam-pointed domain containing Ets transcription factor (SPDEF) [73] was significantly increased when calves latently infected with BoHV-1 were treated with DEX for 3 h to initiate reactivation from latency. Interestingly, KLF15, Slug, and SPDEF are also expressed in more mouse TG neurons following explant when treated with DEX confirming these cellular transcription factors are part of the stress response [74]. In response to stress, GR and KLF15 regulate gene expression dynamics via a feed-forward loop [75, 76]. The hallmark of this feed-forward loop is GR stimulates KLF15 expression and GR and KLF15 form a stable complex and activate expression of genes in specific pathways, including enhanced expression of amino acid metabolizing enzymes and adipogenesis [75, 76].
GR and Stress-Induced Cellular Transcription Factors Activate ICP0, ICP4, and VP16 Promoter/Regulatory SequencesThe ability of GR and/or stress-induced transcription factors to transactivate promoter/regulatory sequences of the ICP0, ICP4, or VP16 genes was examined in transient transfection studies. The rational for these studies is that ectopic expression of these genes initiates reactivation from latency in TG cultures prepared from latently-infected mice [43]. An ICP0 promoter fragment spanning − 800 to + 150 relative to the transcription initiation site was initially examined (Fig. 2A) because this construct is stimulated by heat stress [77]. GR, KLF15, and DEX treatment cooperatively transactivate the full-length ICP0 promoter in spite of no consensus GREs [78]. Conversely, the other stress-induced transcription factors discussed in “Identification of Stress-Induced Cellular Transcription Factors” section did not have a profound effect. Four cis-regulatory modules (CRMs) that span ICP0 promoter sequences upstream of the TATA box were inserted at the 5’ terminus of a simple promoter that drives luciferase activity (Fig. 2B). All but the CRM C fragment was cooperatively transactivated by GR, KLF15, and DEX in Vero cells. Conversely, GR or KLF15 and DEX are sufficient for transactivation in Neuro-2A cells (Fig. 2B) [79]. Mutagenesis of Sp1 binding sites (GGGCGG or CCGCCC) in fragments A, B, and D reduced transactivation by GR, KLF15, and/or DEX to basal levels. GR and KLF15 occupy ICP0 promoter sequences in transfected cells and early times after infection [
留言 (0)