
記住我
Systemic lupus erythematosus (SLE), mainly affecting women of childbearing age, is a chronic autoimmune disease that involves multiple organs such as the skin, joints, kidneys, and central nervous system (1). The estimated global incidence and prevalence of SLE were 5.14 per 100,000 person-years (1.4 to 15.13) and 43.7 per 100,000 person-years (15.87 to 108.92), respectively (2).
SLE is characterized by immune dysregulation and loss of tolerance to autoantigens (3). Both innate and adaptive immunity are involved in the pathogenesis of SLE (4). Autoreactive B cells which are induced by the breakdown of central and peripheral tolerance mechanisms, occupying a core position in the adaptive immune response of SLE, produce autoantibodies, present autoantigens and activate autoreactive T cells (4, 5). CD4+T helper cell subsets such as Th1, Th2, Th17, T follicular helper and regulatory T cells exist disturbances in number and function. The reduced cytolytic activity of CD8+ cytotoxic T cells induces higher rates of infection and sustenance of autoimmunity (6). The population of clusters of double-negative T cells CD4-CD8- T cells, which can produce inflammatory cytokines and infiltrate target tissues, is increased in SLE patients (7, 8). In the innate immune response, abnormal activation or disabled tolerance of dendritic cells induces abnormal production of type I interferons and inflammatory mediators, which contributes to pathogenic innate immunity and autoinflammation (9). In SLE, the defective clearance of apoptotic material and neutrophil extracellular traps lead to the exposure of autoantigens and trigger the production of autoantibodies (10, 11). Local depositions of the immune complexes that are generated between autoantibodies and autoantigens result in serious inflammation through activating the complement system (12, 13).
The etiology of SLE is still not entirely clear. It is now believed that genetic and environmental factors mainly contribute to the development of SLE (14). The genome-wide association study has identified more than 60 risk loci for SLE susceptibility (15). Individuals with identified genetic polymorphisms have a higher risk of developing SLE than the general population (16). Environmental factors including ultraviolet light (17), silica exposure (18), cigarette smoking (19), viral and bacterial infections (20), and sex hormones (21) are involved in the pathogenesis of SLE. Currently, the main therapeutic drugs for SLE are glucocorticoids, immunosuppressive drugs and antimalarial drugs, but their use is limited due to serious side effects (22, 23). Gut microbiota connects some external environmental effectors with the immune system and supports the immune system program to tolerate innocent external and self-antigens (24). However, when the gut microbiota is dysbiotic, it can disrupt immune function, induce inflammation and immune system sensitization and lead to autoimmune diseases (25, 26). Recently, increasing studies have found that gut microbiota as an environmental factor contributes to the development of SLE (27), and modulating the gut microbiota appears to be a potential treatment for SLE (28). In this review, we summarize the alteration of gut microbiota in SLE, potential mechanisms connecting gut microbiota to SLE, and immune dysregulation related to gut microbiota in SLE.
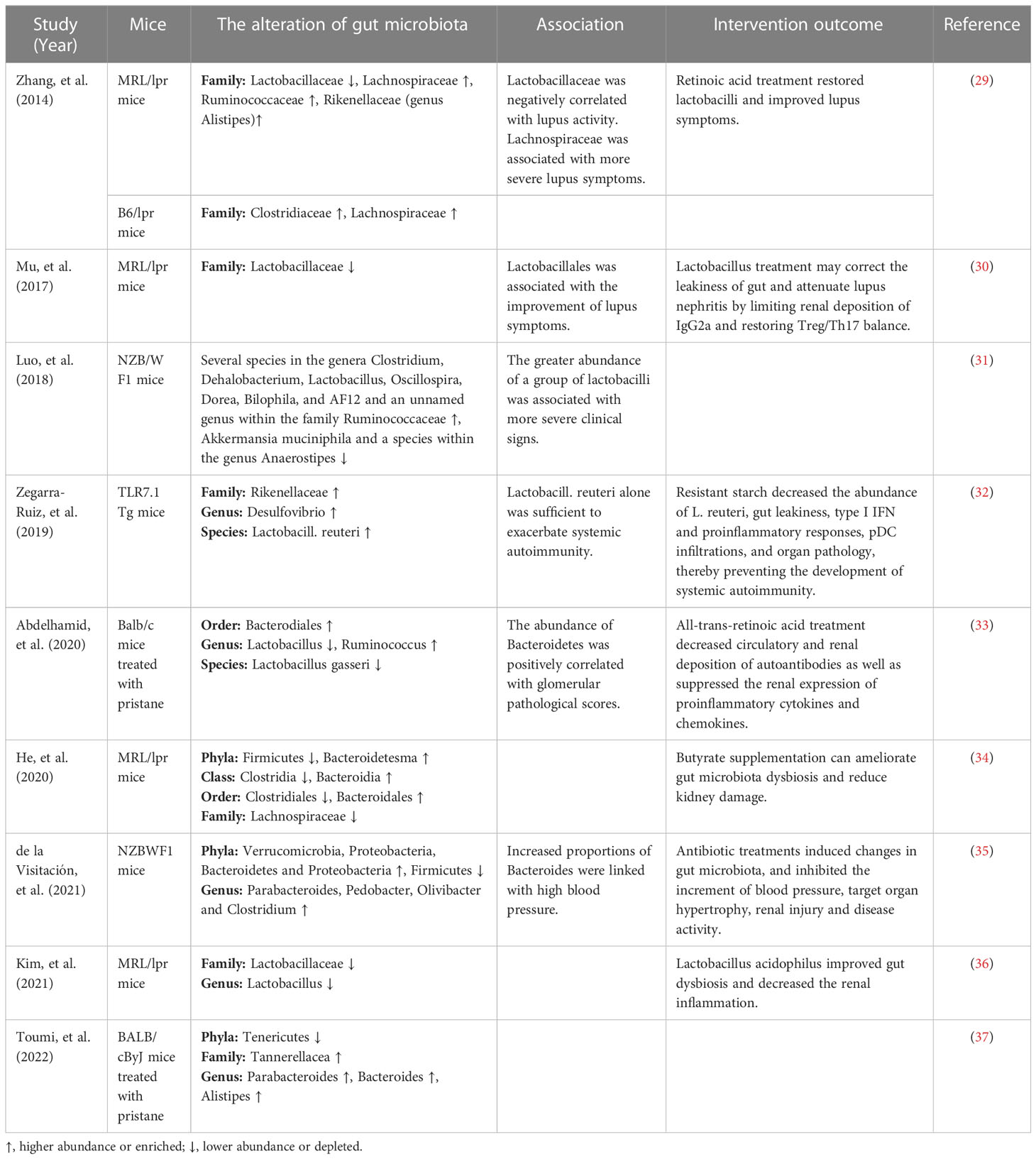
The alteration of gut microbiota in SLEThe changes of gut microbiota in lupus mice modelsRecently, increasing studies have revealed that dysbiosis of gut microbiota is associated with SLE. Alteration of the gut microbiota has been found in various lupus mouse models, as shown in Table 1. Luo et al. (31) found that the gut microbiota changed significantly before and after lupus onset in NZB/W F1 mice. Zhang et al. (29) found that Lactobacillaceae significantly decreased and Lachnospiraceae significantly increased in lupus-prone MRL/lpr mice. The decreased abundance of Lactobacillaceae was also observed in MRL/lpr mice in another study (36). Increased intestinal abundance of Lactobacillaceae was associated with improvement of lupus symptoms, while increased colonization of Lachnospiraceae was associated with disease progression (29). Consistently, Mu et al. (30) found that Lactobacillales significantly depleted in the gut microbiota of MRL/lpr mice and was associated with improvement of lupus symptoms. However, in another lupus mice model NZB/W F1 mice, Luo et al. (31) reported that the greater abundance of a group of Lactobacilli was linked with more severe clinical signs. Zegarra-Ruiz et al. (32) found that the abundance of Lactobacill. reuteri increased in TLR7.1 Tg mice and Lactobacill. reuteri colonization exacerbated systemic autoimmunity under specific-pathogen-free and gnotobiotic conditions. In addition, He et al. (34) observed a rise in the abundance of Bacteroidetes and a reduction in Firmicutes in the gut microbiota of MRL/lpr mice. Abundant Bacteroidetes and decreased Firmicutes were also found in NZBWF1 mice, and increased proportions of Bacteroides were associated with high blood pressure (35). Abdelhamid et al. (33) found that the abundance of Bacteroidetes was positively correlated with glomerular pathological scores. A lower Firmicutes/Bacteroidetes(F/B) ratio found in 6-week-old MRL/lpr mice might play an important role in promoting early disease onset (38). Furthermore, Chen et al. (39) reported that the increased abundance of genera Candidatus saccharimonas, Desulfovibrio, Odoribacter and Roseburia in mice treated with HCMVpp65 peptide is significantly correlated with lupus-like effects including enhanced levels of creatinine, proteinuria, glomerular damage and anti-dsDNA antibodies. Valiente et al. (40) found that NZM2410 mice colonized with segmented filamentous bacteria showed worsening glomerulonephritis, deposition of glomerular and tubular immune complexes, and interstitial inflammation. Therefore, gut microbiota dysbiosis in lupus mice models could be characterized by a decrease of beneficial bacteria and an increase of harmful bacteria and was associated with SLE.
Table 1 The alteration of gut microbiota in lupus mice models.
The gut microbiota from lupus patients and mice in recipient mice can cause the production of autoantibodies and increase the expression of lupus-related genes. Ma et al. (41) conducted a study transplanting fecal microbiota from SLE mice to germ free mice and found that fecal microbiome from SLE mice could induce the production of anti-dsDNA antibodies and upregulate the expression of SLE susceptibility genes in germ free mice. Choi et al. (42) transferred the dysbiotic gut microbiota from triple congenic lupus-prone mice into germfree congenic C57BL/6 mice, and found that the gut microbiota activated immune cells and induced the production of autoantibodies in recipient mice. In a study by Ma et al., when receiving fecal microbiota from SLE patients, germ-free mice showed increased expression of genes associated with SLE and a range of lupus-like phenotypic features, including imbalanced cytokines, elevated serum levels of autoimmune antibodies, changed the distribution of immune cells in mucosal and peripheral immune responses (43). In addition, high blood pressure and vascular complications were found in germ-free or germ-depleted mice after receiving gut microbiota from hypertensive NZBWF1 mice (35). These results further illustrate that dysbiosis of gut microbiota contributes to the development of SLE.
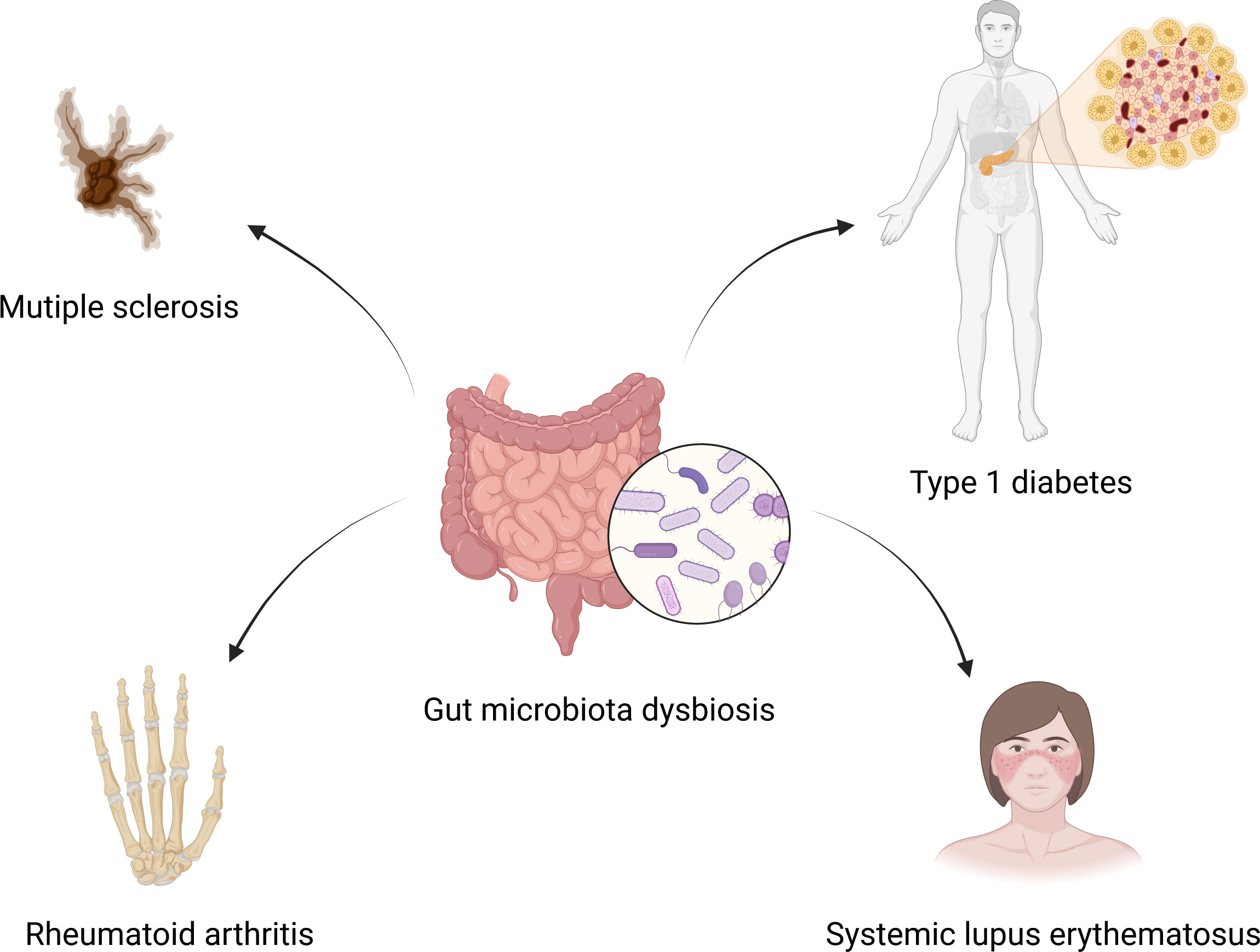
The changes of gut microbiota in SLE patientsIt has been reported that patients with extra-intestinal autoimmune diseases, including multiple sclerosis (44), rheumatoid arthritis (45), and type 1 diabetes (46), have distinct gut microbiota compositions compared to healthy individuals. Plenty of studies have also shown that gut microbiota altered significantly in SLE patients compared to healthy controls. Extra-intestinal autoimmune diseases associated with gut microbiota dysbiosis are summarized in Figure 1. Hevia et al. (47) performed a study comparing the fecal microbial profiles between SLE patients and healthy subjects and published the first report to describe an SLE-associated intestinal dysbiosis in humans. Wang et al. (48) compared lupus patients to their healthy family members, controlling for living conditions and dietary factors, and found that the gut microbiota of lupus patients still differed from that of healthy controls. It is known that Bacteroidetes and Firmicutes are the most abundant components of the human gut microbiota (49). A significantly lower F/B ratio was observed in SLE patients compared to healthy subjects (47). Several subsequent studies conducted in different countries around the world have also observed a lower F/B ratio in the gut microbiota of lupus patients compared to healthy people (37, 50–52). Furthermore, Katz-Agranov and Zandman-Goddard (53) revealed that SLE individuals even in remission had a significantly lower F/B ratio. Active SLE patients showed a significantly lower F/B ratio compared to the inactive SLE group (37). In addition, Gerges et al. (52) found that the F/B ratio was negatively correlated with the SLEDAI-2K score. Widhani et al. (54) also reported that SLE patients with mild disease activity had a higher F/B ratio compared to patients with moderate or high disease activity. These results indicate that the lower F/B ratio in SLE patients is independent of ethnicity, lifestyle as well as disease stage and inversely correlated with lupus disease activity. However, a lower F/B ratio is not the specific feature in the alteration of gut microbiota in SLE patients, and it is also linked to other diseases, such as type 2 diabetes (55), Crohn’s disease (56) and Parkinson’s disease (57). Reduced bacterial diversity is another main feature of intestinal dysbiosis that has been reported by many studies conducted in humans (31, 51, 58–60). Furthermore, patients with a high SLE activity index had a particularly significant reduction in the species diversity of gut microbiota (58). In addition, a recent study reported that the gut microbiota composition of SLE patients with depression was also different from that of SLE patients without depression (61).
Figure 1 Gut microbiota dysbiosis and extra-intestinal autoimmune diseases. Gut microbiota dysbiosis is associated with multiple sclerosis, rheumatoid arthritis, type 1 diabetes and systemic lupus erythematosus. Created by BioRender.com.
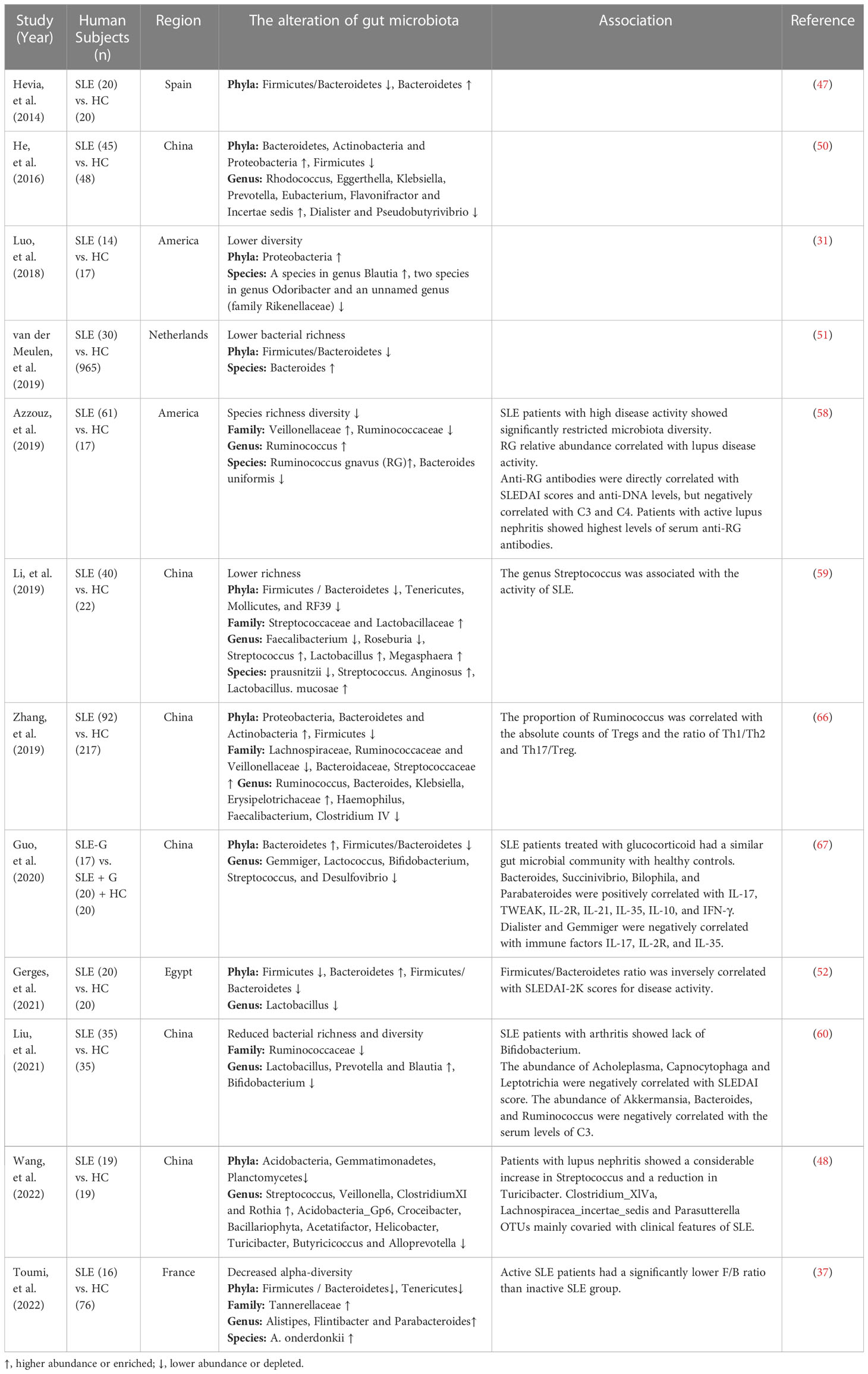
The indicators commonly used to reflect the disease activity of SLE patients include SLEDAI, anti-double stranded DNA (anti-dsDNA), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP) and complement C3 (62–64). Some biomarkers in the gut microbiota of SLE patients have been found to be associated with the disease activity indicators. Bagavant et al. (65) found that higher titers of anti-E. gallinarum IgG in patients were significantly correlated with the presence of anti-Ribosomal P, anti-dsDNA and anti-Sm autoantibodies. Azzouz et al. (58) reported that Ruminococcus gnavus (RG) showed a mean 5-fold overabundance in lupus patients compared with healthy controls. And patients with high disease activity and especially lupus nephritis showed the greatest expansion of RG. Furthermore, anti-RG antibodies were directly correlated with anti-DNA levels and SLEDAI scores, while they were negatively correlated with C3 and C4. Patients with active nephritis including Class III and IV exhibited the highest levels of anti-RG strain restricted antibodies in serum. The abundance of Acholeplasma, Capnocytophaga and Leptotrichia were negatively correlated with SLEDAI score, and the abundance of Bacteroides, Ruminococcus and Akkermansia had an inverse correlation with the serum levels of complement C3 (60). The genus Streptococcus was associated with the lupus activity (59). These findings suggest that some biomarkers in the gut microbiota are linked with the disease activity of SLE patients and have the potential to be used as indicators to reflect the disease activity of SLE patients. Studies on the gut microbiota in lupus patients are summarized in Table 2.
Table 2 The alteration of gut microbiota in SLE patients.
In recent years, studies have further confirmed the association between gut microbiota and SLE, and pointed out a causal relationship between the two. Xiang et al. (68) performed a meta-analysis including 11 case-control studies conducted in five countries and nine cities, and found increased abundance in Enterobacteriaceae and Enterococcaceae and decreased abundance in Ruminococcaceae in the gut microbiota of patients with SLE. This study added to evidence that dysbiosis of gut microbiota is present in lupus patients. Furthermore, a two-sample mendelian randomization study found that Actinobacteria, Bacillales, Coprobacter and Lachnospira were inversely correlated with the risk of SLE, and Bacilli, Eggertella and Lactobacillales might be the risk factors of SLE. More importantly, this study showed causal effects of gut microbiota on SLE (69).
Potential mechanisms linking gut microbiota to SLEImpaired intestinal barrier and pathogen translocationThe intestinal epithelium not only separates the host from the external environment, but also acts as a first-line innate immune defense against the entry of foreign antigens. A normal gut barrier has size selectivity to prevent the translocation of viable organisms and several PAMPs, which can trigger the systemic inflammation (70). When the intestinal barrier is compromised, pathogens can translocate to systemic circulation and internal organs. It has been reported that the gut microbiota and leaky gut are related to autoimmune diseases including type 1 diabetes (71) and multiple sclerosis (72). Many studies as follow have revealed that intestinal barrier impairment and pathogen translocation also occurred in lupus mice and patients.
In lupus mice models, Mu et al. (30) found that endotoxin levels were significantly higher in the blood of MRL/lpr mice compared to MRL/MP controls. MRL/Lpr mice had significantly abundant FITC-dextran in the blood when gavaged with FITC-dextran compared to MRL/MP mice. Vieira et al. (73) reported that fluorescein isothiocyanate (FITC)– dextran was found in the systemic circulation of (NZW × BXSB)F1 hybrid mice after being taken orally. E. gallinarum was detected in the mesenteric veins, mesenteric lymph nodes (MLN), spleen, and liver of (NZW × BXSB)F1 hybrid mice. Both TLR7.1 Tg and C57Bl/6 mice treated with imiquimod showed impaired gut barrier, which was reflected by leakage of FITC-dextran into the systemic circulation. L. reuteri was found to translate into the mesenteric lymph nodes (MLN), spleen, and liver of TLR7.1 Tg mice (32). Toral et al. (74) observed significantly higher endotoxin levels in plasma in NZBWF1 mice compared with the control group.
In healthy people, an intact intestinal barrier can prevent luminal contents and systemic IgG from leaking into the gut lumen. Calcitonin is a recognized biomarker of intestinal barrier deficiency (58). In SLE patients, increased fecal albumin and calprotectin were found in the stool sample (73). Consistently, Azzouz et al. (58) also observed an increase in IgG and a higher level of calprotectin in the fecal sample of SLE patients. These results suggested impaired gut barrier function in SLE patients. E. gallinarum was detected in liver biopsy samples from both lupus patients and autoimmune hepatitis patients (73). Elevated serum soluble CD14 and α1-acid glycoprotein levels were found in lupus patients, which indicated translocation of the gut microbiota (58).
Gut-leakage is also associated with lupus disease progression. Thim-Uam et al. (75) used dextran sulfate solution (DSS) to induce gut-leakage in FcGRIIb-/- mice and found that DSS-induced gut-leakage induced high anti-dsDNA immunoglobulin in serum, enhanced lupus features including proteinuria and serum creatinine, caused the gut translocation of molecular components of gut pathogens, enhanced MLN apoptosis and induced spleen apoptosis in FcGRIIb-/- mice. Silverman et al. (76) colonized C57BL/6 mice with individual Ruminococcus gnavus (RG) strains from lupus patients, and found that lupus-derived RG strains enhanced intestinal permeability, elevated serum levels of zonulin which is a regulator of tight junction formation between cells forming the intestinal barrier, and translocated to mesenteric lymph nodes. The level of intestinal permeability induced by RG has a significant correlation with anti-native DNA autoantibodies and serum IgG anti RG cell-wall lipoglycan antibodies.
Interestingly, the “leakiness” of the gut epithelium could be reduced or even reversed, and the translation of gut bacteria could be suppressed. Antibiotics reduced intestinal leakage and suppressed microbial translocation (73, 77). Intramuscular vaccination against E. gallinarum prevented E. gallinarum translocation into internal organs (73). Resistant starch tightened the gut epithelial barrier and reduced the translocation of L. reuteri (32). Lactobacillus supplementation enhanced the barrier function of the intestinal epithelium (74). All-trans-retinoic acid treatment reversed pristane-induced leaky gut (33). Treatment with larazotide acetate, a specific molecular antagonist of zonulin, completely reversed gut permeability (76).
In summary, intestinal microbiota translocates to systemic circulation and internal organs since the intestinal barrier is compromised, increases exposure to autoantigens through inducing or enhancing apoptosis of cells such as splenocytes, and ultimately leads to increased deposition of immune complexes in organs then aggravates lupus. Interestingly, some interventions can reduce or even reverse the intestinal leakage to alleviate lupus.
Molecular MimicryMolecular mimicry is another potential mechanism linking gut microbiota to SLE. Gut microbiota can mimic autoantigens through their proteins and metabolites (78). In hosts carrying high risk human leukocyte antigen (HLA) genes, continued colonization of them with bacteria expressing cross-reactive epitopes may continuously activate cross-reactive autoreactive T cells in the gut, especially in the case of impaired intestinal barrier (79). Patients with SLE produce autoantibodies against Ro60 which is an evolutionarily conserved RNA binding protein, and the antibody against Ro60 is the most common and earliest preclinical anti-nuclear antibody (80). Greiling et al. (80) found that bacteria expressing Ro60 orthologs was present in the skin, oral, and gut of both lupus patients and healthy controls. Sera from SLE patients with positive anti-Ro60 immunoprecipitated commensal Ro60 ribonucleoproteins, and Ro60-containing bacteria from skin and activated human Ro60 autoantigen-specific CD4 memory T cell clones. Furthermore, colonization of germ-free mice with Bacteroides thetaiotaomicron containing Ro60 ortholog caused T and B cell responses against human Ro60 and glomerular immune complex deposition (80).
Anti-β2GP1 antibody is also a diagnostic index for SLE. β2GP1 ortholog expressed by E. gallinarum was found to induce anti-β2GP1 antibody (73). Ruff et al. (81) found that β2GPI-reactive memory CD4+ memory T cell clones and APS-derived β2GPI autoantibody cross-reacted with mimotopes expressed by a gut commensal Roseburia intestinalis. Moreover, oral gavage of (NZW x BXSB)F1 mice with Roseburia intestinalis led to increased anti-human β2GPI IgG autoantibodies and thrombotic events.
In addition, Azzouz et al. (58) found that antigen in Ruminococcus gnavus strain CC55_001C could cross-react with anti-dsDNA antibodies. The peptide “YLYDGRIFI” similar to human Sm antigen epitope from Odoribacter splanchnicus increased secretion of IFN-γ and IL-17A. And the peptide “DGQFCM” mimicking human Fas antigen from Akkermansia muciniphila specifically binds to the IgG produced by memory B cells from lupus patients (27).
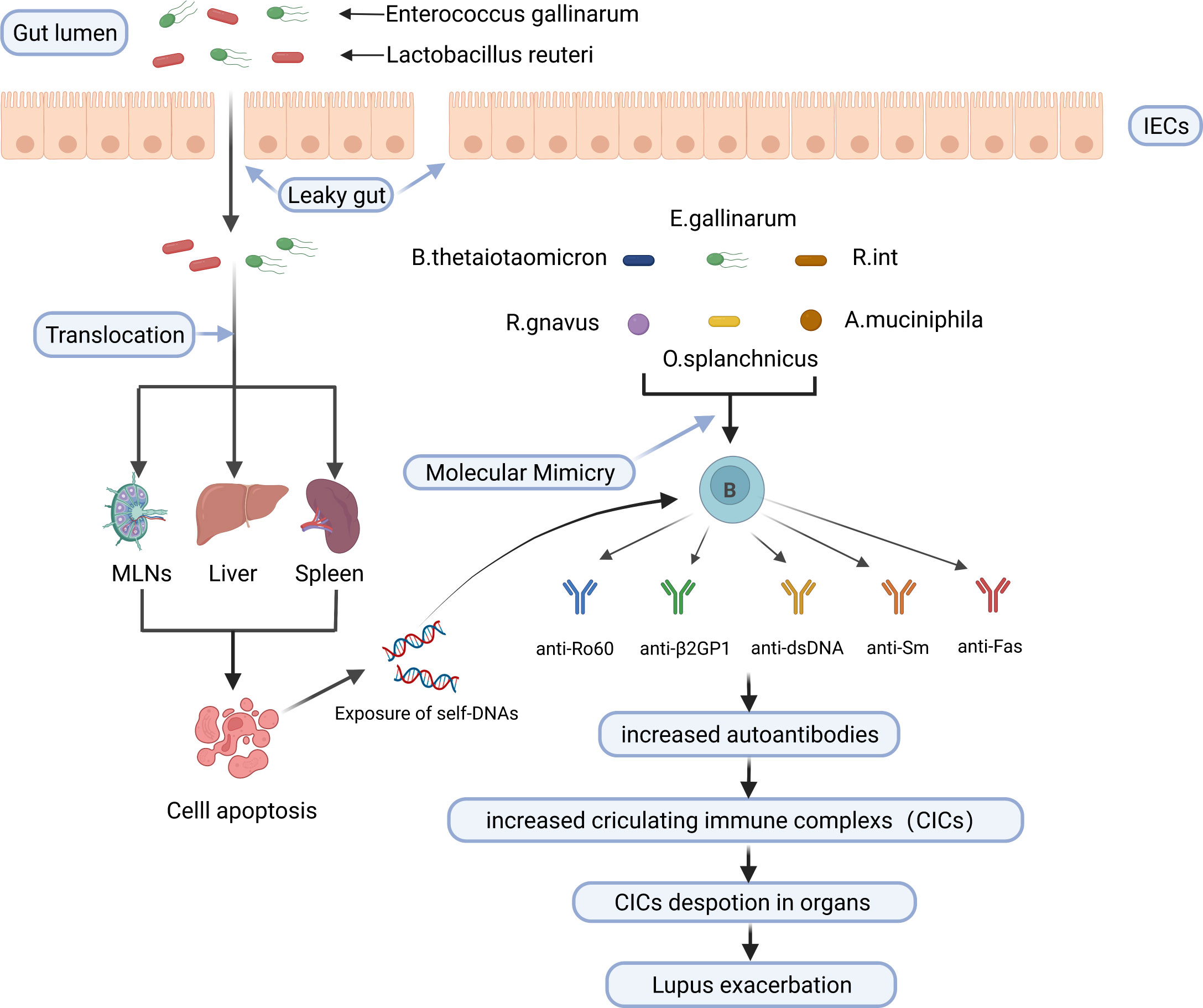
In conclusion, gut microbiota activates autoreactive T and B cells and triggers autoimmunity through encoding autoantigen orthologues and non-orthologous mimotopes of autoantigens, thus contributing to the development of SLE. Potential mechanisms linking the gut microbiota to SLE are shown in Figure 2.
Figure 2 Potential mechanisms linking the gut microbiota to SLE. E. gallinarum and L. reuteri can translocate to internal tissues and organs such as mesenteric lymph nodes (MLN), liver, and spleen in the case of impaired gut barrier function, increase exposure to autoantigens through inducing or enhancing apoptosis of cells such as splenocytes, and increase the production of autoantibodies. B. thetaiotaomicron, E. gallinarum, R. int, R. gnavus, O. splanchnicus and A. muciniphila trigger cross-reactive B cell response through molecular mimicry of human autoantigens by their bacterial orthologs. Finally, increased circulating immune complexs deposit in organs and exacerbate lupus. IECs, intestinal epithelial cells; MLNs, mesenteric lymph nodes; B. thetaiotaomicron, Bacteroides thetaiotaomicron; R. int, Roseburia intestinalis; R. gnavus, Ruminococcus gnavus; O. splanchnicus, Odoribacter splanchnicus; A. muciniphila, Akkermansia muciniphila. Created by BioRender.com.
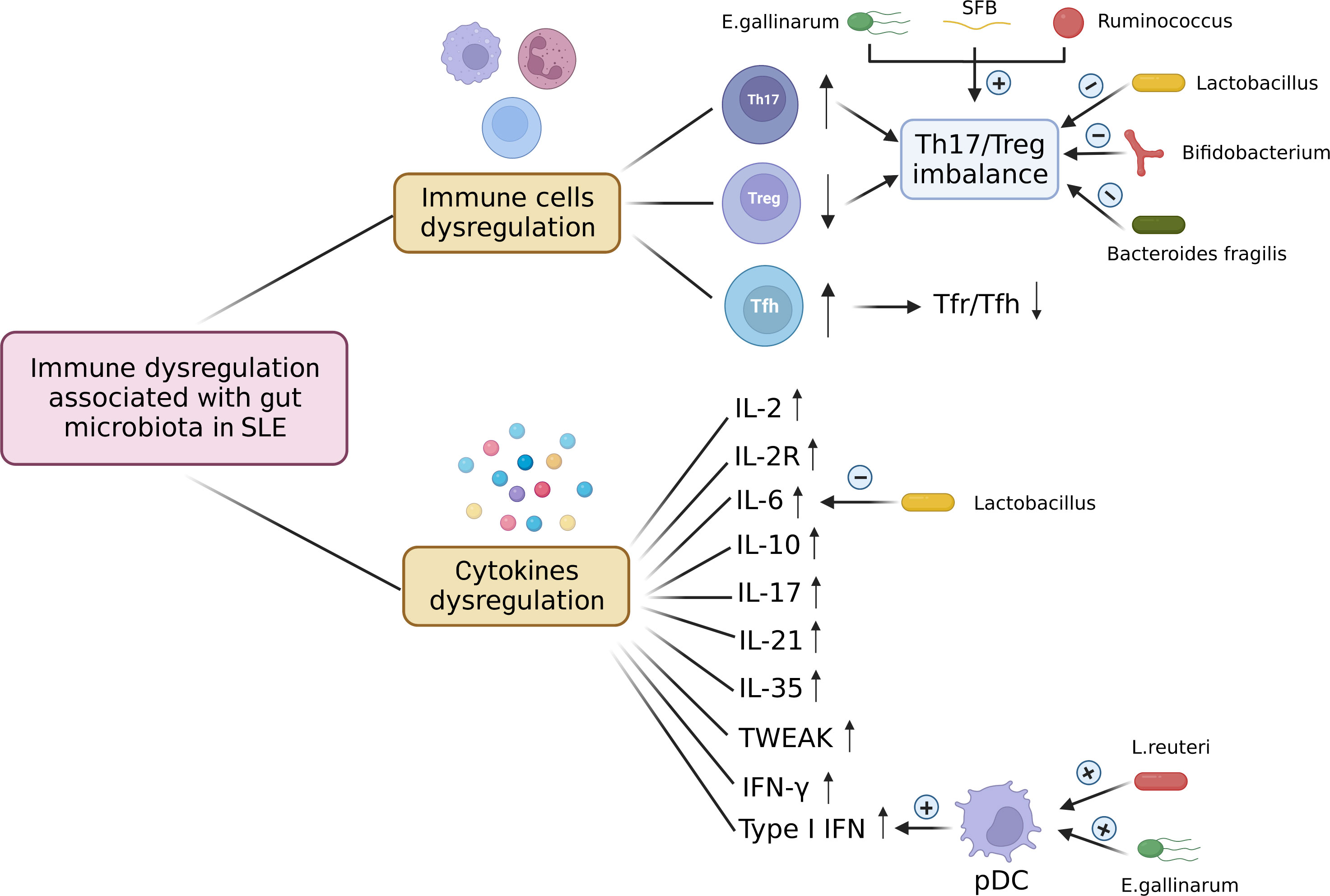
Immune dysregulation associated with gut microbiota in SLEImmune cells dysregulationThe imbalance between anti-inflammatory regulatory T(Treg) and inflammatory T-helper 17 (Th17) cells plays a significant role in the pathogenesis of SLE (82, 83). Recently, some studies have revealed that some specific pathogens contributed to Th17/Treg imbalance in lupus mice models. Vieira et al. (73) found that Th17 cells were induced in the small intestinal lamina propria and mesenteric lymph nodes of C57BL/6 mice monocolonized with E. gallinarum. The induction of Th17 and autoantibodies by E. gallinarum was eliminated by the administration of a selective AhR antagonist, which indicates that E. gallinarum promotes autoimmunity by AhR signaling. Zhang et al. (66) reported that the proportion of Ruminococcus was associated with the absolute counts of Treg cells and the ratio of Th17/Treg and Th1/Th2. NZM2410 mice colonized with segmented filamentous bacteria (SFB) showed increased Th17 cells in small intestinal lamina propria (40). Besides, the intestinal microbiota from SLE patients or mice also induced Th17/Treg imbalance in germ-free mice. Germ-free mice treated with feces from SLE mice showed increased B cells and significantly less abundant Treg cells mice in intestinal mucosa compared to those treated with feces from B6 mice (41). Ma et al. (43) found an increase in the frequency of Th17 cells and a reduction in Treg cells in the spleen of germ-free mice gavaged with feces from SLE patients. Additionally, the frequency of Tfh cells in circulation (84) and the Tfh/Tfr ratio (85) are positively correlated with lupus activity. Choi et al. (42) found that germ-free B6 mice that received feces from triple congenic lupus-prone mice exhibited an increased frequency of Tfh cells and a decreased ratio of Tfr to Tfh.
Interestingly, probiotic treatment could restore the balance between Treg cells and Th17 cells. Bifidobacterium can maintain the balance of Treg/Th17/Th1 by suppressing the excessive activation of CD4+ lymphocytes in SLE patients (86). Lactobacillus supplement could increase Treg cells and reduce Th17 cells to restore Th17/Treg balance (30, 74, 87). Bacteroides fragilis treatment restored the Th17/Treg balance and ameliorated the lupus activity of MRL/lpr mice (88). Lactobacillus fermentum CECT5716 and Bifidobacterium breve CECT7263 treatment restored the Th17/Treg balance in MLNs and reduced vascular Th1, and Th17 infiltration to restore endothelial function in a mouse lupus model induced by activating TLR-7 (89). Furthermore, Kim et al. (36) found that Lactobacillus acidophilus modulated Th17/Treg balance in MRL/lpr mice by the SIGNR3 pathway. In addition, regulated B (Breg) cells can exert immunosuppressive effects and support immune tolerance (90). Mu et al. (91) reported that oral administration of bacterial DNA could induce Breg cells and alleviate lupus.
Cytokines dysregulationIn a series of immune-mediated diseases, cytokines act as critical mediators of inflammation and tissue damage (92). Many studies have revealed that T cells produced cytokines abnormally in SLE patients (93). IL-6, a pro-inflammatory cytokine produced by activated antigen-presenting cells and T cells, is known to promote B cells to produce antibodies (94) and suppress Treg cells (95). IL-17 is dysfunctional in SLE and promotes the disease progression (96). A meta-analysis displayed that the level of IL-17 has a positive correlation with lupus activity (97). IL-10 can inhibit kidney disease by inhibiting IFNγ-mediated production of IgG2a, which is a major immune deposit in the kidney of MRL/lpr mice (98). Mu et al. (30) found that Lactobacillus treatment decreased IL-6 and increased IL-10 production in the gut contributing to an anti-inflammatory environment. In SLE patients, twelve cytokines including IFN-γ, IL-1β, IL-2R, IL-6, IL-8, IL-10, IL-17, IL-21, IL-22, IL-35, TNF-α and TWEAK displayed higher expression levels compared to healthy controls. Bacteroides, Bilophila, Parabateroides, and Succinivibrio were positively correlated with IL-2R, IL-10, IL-17, IL-21, IL-35, TWEAK, and IFN-γ. Dialister and Gemmiger had a negative correlation with IL-2R, IL-17, and IL-35 (67). Yao et al. (61) also reported that SLE patients displayed higher serum levels of IL-2 and IL-6. The abundance of Roseburia and Faecalibacterium was inversely correlated with IL-6, the abundance of Roseburia had a negative correlation with IL-2, and the abundance of Bacteroides had a positive correlation with IL-2. Therefore, gut microbiota dysbiosis is associated with dysregulated cytokines in SLE. Interestingly, SLE patients treated with synbiotics had a significant decrease in serum IL-6 (54). Type I IFN, primarily produced by plasmacytoid dendritic cells, is a major pathogenic factor in SLE (99, 100). Vieira et al. (73) found that the presence of E. gallinarum upregulated Enpp3, which can increase the number of plasmacytoid dendritic cells (pDCs). Both murine hepatocytes and human hepatocytes induced type I interferon under the stimulation of E. gallinarum. Under specific-pathogen-free and gnotobiotic conditions, L. reuteri colonization increased plasmacytoid dendritic cells and interferon signaling (32).
To sum up, gut microbiota dysbiosis contributes to the immune dysregulation in SLE, the immune dysregulation associated with gut microbiota in SLE occurs not only at the immune cell level but also at the cytokine level. The immune dysregulation associated with gut microbiota in SLE is shown in Figure 3.
Figure 3 Immune dysregulation associated with gut microbiota in SLE. The immune dysregulation associated with gut microbiota in SLE occurs not only at the immune cell level but also at the cytokine level. Immune cells dysregulation includes increased Th17 cells, decreased Treg cells, and increased Tfh cells, which leads to the Th17/Treg imbalance and the decreased ratio of Tfr to Tfh. E. gallinarum, SFB, and Ruminococcus induce Th17/Treg imbalance by increasing Th17 cells and decreasing Treg cells. Lactobacillus, Bifidobacterium and Bacteroides fragilis can restore the Th17/Treg balance. The levels of cytokines associated with gut microbiota IL-2, IL-2R, IL-6, IL-10, IL-17, IL-21, IL-35, TWEAK, IFN-γ, and type I IFN are up-regulated in SLE. E. gallinarum and L. reuteri increase the number of plasmacytoid dendritic cells (pDCs) and promote the expression of type 1 interferon. Lactobacillus treatment can decrease the level of IL-6. pDC, plasmacytoid dendritic cell; SFB, segmented filamentous bacteria; E. gallinarum, Enterococcus gallinarum; L. reuteri, Lactobacillus reuteri. Created by BioRender.com.
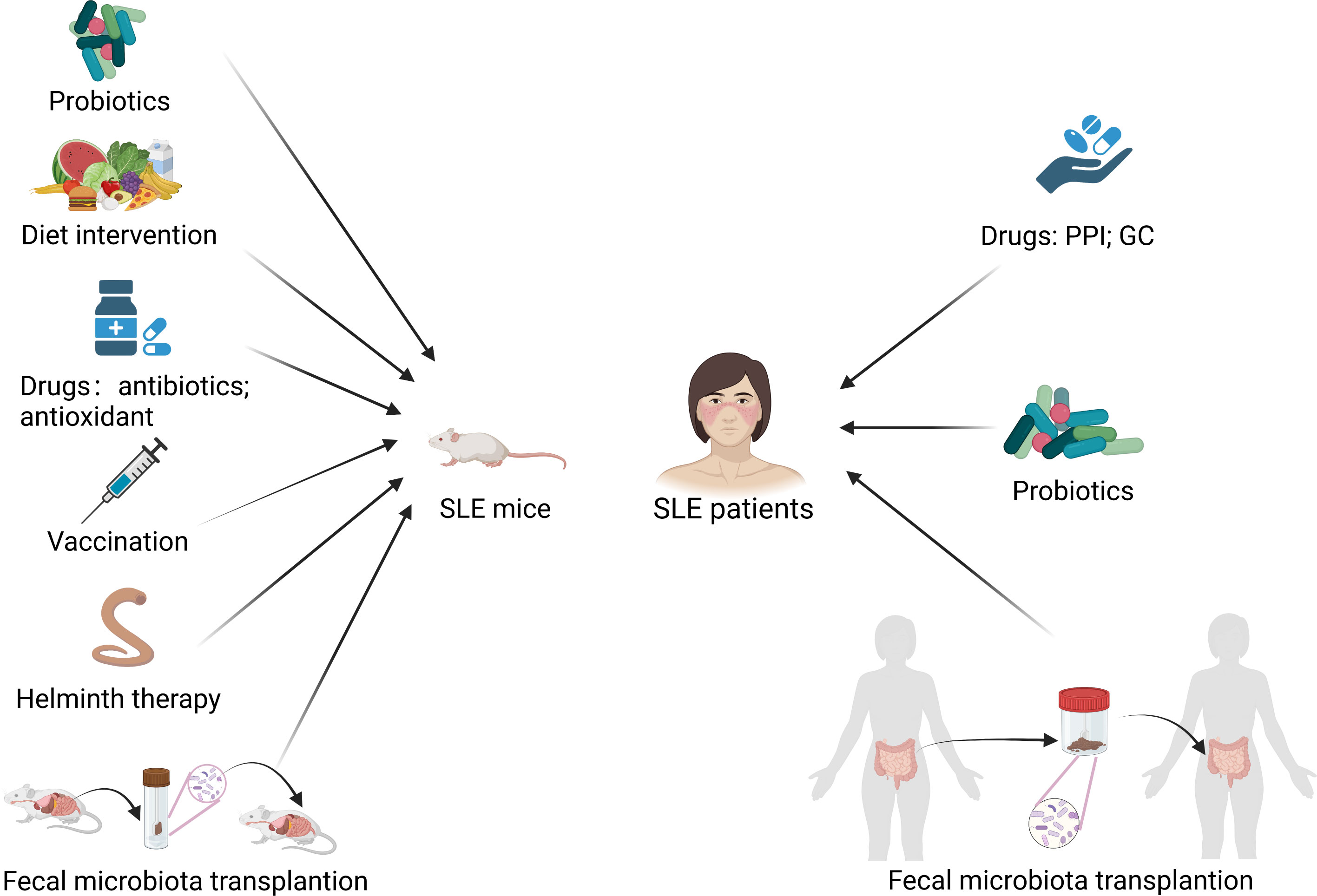
Gut microbiota: potential diagnostic and therapeutic value for SLEGut microbiota modulation to treat lupus mice modelsLactobacillus, known as probiotic, plays an anti-inflammatory role in autoimmune disease (101). Many studies have shown the beneficial effects of Lactobacilli supplementation in SLE mice. Hsu et al. (102) found that supplementary treatment of Lactobacillus paracasei GMNL-32, Lactobacillus reuteri GMNL-89 and L. reuteri GMNL-263 mitigated hepatic inflammation and apoptosis in NZB/W F1 lupus-prone mice. Lactobacillus treatment attenuated lupus nephritis through reducing renal deposition of IgG2a in MRL/lpr mice (30). NZBWF1 mice exhibited reductions in blood pressure, cardiac and renal hypertrophy, splenomegaly and lupus activity after Lactobacillus fermentum CECT5716 treatment (74). Manirarora et al. (87) reported that feeding Lactobacilli might delay lupus progression in BWF1 mice. Lactobacillus acidophilus improved gut dysbiosis, decreased the renal inflammation and enhanced the therapeutic effect of tacrolimus in MRL/lpr mice. However, Luo. et al. (31) found that the greater abundance of a group of Lactobacilli in NZB/W F1 mice might be linked to more severe disease. Lactobacillus reuteri can drive autoimmunity in TLR7-dependent mouse models of SLE (32). Lactobacillus have different effects on various lupus mice models which represent different genetic or environmental conditions, which may indicate that probiotic supplementation in the treatment of lupus should be individualized based on genetic and environmental factors.
Some other interventions such as dietary interventions, drug therapy, vaccination, helminth therapy and fecal microbiota transplantation have also been shown to modulate gut microbiota and exert beneficial effects in lupus mice. Zhang et al. (29) found that retinoic acid treatment restored downregulated Lactobacilli and improved lupus symptoms in lupus-prone mice. All-trans-retinoic acid reduced circulatory and renal deposition of autoantibodies and suppressed the expression of proinflammatory cytokines and chemokines in kidney in Balb/c mice treated with pristane (33). Resistant starch decreased the abundance of Lactobacill. reuteri and prevented the development of systemic autoimmunity in TLR7.1 Tg mice (32). Johnson et al. (103) treated (SWR×NZB) F1 (SNF1) mice with acidic pH water and neutral pH water respectively, and found that the composition of gut microbiota is significantly different between two groups of mice. Mice treated with acidic pH water developed nephritis at a slower pace than those treated with neutral pH water. Low dietary tryptophan has been reported to prevented autoimmune pathology in lupus-prone mice (42). He et al. (34) found that butyrate supplementation could ameliorate gut microbiota dysbiosis and reduce kidney damage in MRL/lpr mice. Mu et al. (77) found that antibiotics treatment reshaped the gut microbiota by reducing potentially harmful bacteria and enriching potentially beneficial bacteria and ameliorated systemic autoimmunity as well as kidney histopathology in MRL/lpr mice. In NZBWF1 mice, antibiotics was also found to change the composition of gut microbiota, suppress the elevation of blood pressure, and reduce renal injury and disease activity (35). Antioxidant N-acetylcysteine altered the composition of gut microbiota and alleviated autoimmunity in MRL/lpr mice (38). (NZW × BXSB) F1 mice showed restored intestinal barrier function and alleviated lupus after being inoculated with the Enterococcus gallinarum vaccine (73). It was reported that gastrointestinal helminth infection could modulate the gut microbiota (104). Olia et al. (105) found that infection with Hymenolepis microstoma inhibited NZBWF1 mice from developing lupus symptoms including production of autoantibody, proteinuria, glomerular histopathology, and splenomegaly. Wang et al. (106) transferred the gut microbiota of MRL/lpr mice treated with prednisone into the blank MRL/lpr mice, and found that prednisone-regulated gut microbiota alleviated lupus but didn’t show side effects as prednisone in MRL/lpr mice. And prednisone-regulated gut microbiota might alleviate lupus by retaining the abundance of Lactobacillus and decreasing Ruminococcus and Alistipes. Therefore, interventions which can modulate the dysbiosis of gut microbiota, such as dietary interventions, drug therapy, vaccination, helminth therapy and fecal microbiota transplantation, could be potential treatments for lupus patients.
Gut microbiota modulation to treat SLE patientsDrugs can also affect the gut microbiota of lupus patients. Some studies have reported that the altered gut microbiota in lupus patients treated with some drugs becomes similar to the gut microbiota of normal individuals. Guo et al. (67) also found a lower Firmicutes/Bacteroidetes ratio in SLE patients, and the ratio increased in those having undergone glucocorticoid treatment. Besides, patients treated with glucocorticoids had similar gut microbial community to healthy controls, which means that changes in the gut microbiome might represent a return to homeostasis. Li et al. (107) reported that the gut microbiota of SLE patients treated with proton pump inhibitors (PPIs) showed increased alpha-diversity and the alpha-diversity became similar to healthy controls. PPIs use is related to the increased abundance of beneficial commensals and the decreased abundance of certain opportunistic pathogenic genera such as Escherichia.
Furthermore, several studies have also attempted to treat SLE patients by modulating gut microbiota. Huang et al. (28) conducted the first fecal microbiota transplantation (FMT) clinical trial in active SLE patients by oral encapsulated fecal microbiome from healthy donors, and found that FMT treatment significantly reduced the SLEDAI-2K score and the level of serum anti-dsDNA antibody. They also observed a marked increase in SCFAs-producing bacterial taxa and a decrease in inflammation-related bacterial taxa, with CD4+ memory/naïve ratio and levels of IL-6 decreasing in the peripheral blood and production of SCFAs increasing in the gut after FMT. It was reported that SCFAs played a beneficial immune regulatory role in SLE (108). Widhani et al. (54) found that the supplementation of synbiotics, a combination of prebiotics and probiotics, suppressed the increase of hs-CRP, reduced IL-6 expression, increased the Firmicutes/Bacteroidetes ratio, and improved SLE disease activity index 2K score in SLE patients.
In addition, Li et al. (59) found that the random forest model could distinguish SLE patients from rheumatoid arthritis patients and healthy controls and predict the disease activity of SLE patients, which suggests the potential diagnostic value of gut microbiota as a potential biomarker. Therefore, the gut microbiota is not only a potential biomarker for the diagnosis and prognosis of SLE but also a potential target for the treatment of SLE. Interventions that attempt to treat SLE in mice models and humans by modulating the gut microbiota are summarized in Figure 4.
Figure 4 Modulation of gut microbiota treats SLE mice and patients. PPI, proton pump inhibitor; GC, glucocorticoid. Created by BioRender.com.
ConclusionThe alteration in the gut microbiota of lupus mice and SLE patients is characterized by an increase in detrimental bacteria and a decrease in beneficial bacteria. Gut microbiota dysbiosis triggers autoimmunity through the potential mechanisms of translocation and molecular mimicry, leading to immune cells dysregulation (e.g., Th17/Treg imbalance) and cytokines dysregulation (e.g., increased expression of type I interferon), thereby contributing to the development and progression of SLE. Some interventions to modulate gut microbiota, such as dietary intervention, drug therapy (e.g., antibiotic and antioxidant N-acetylcysteine), probiotic supplementation, helminth therapy, vaccination and fecal microbiota transplantation, are potential treatments for SLE. Furthermore, gut microbiota is not only a potential therapeutic target for SLE but also a potential biomarker for the diagnosis and prognosis of SLE. However, there are few studies on the intervention of modulating gut microbiota such as fecal microbiome transplantation in the treatment of SLE patients. More studies are needed to verify the feasibility, safety and effectiveness of this approach.
Author contributionsTBZ contributed to the conception and design of this review. KJY performed most of the overall work for this report and wrote the first manuscript. YNX, JLW, YDL and XTC reviewed and checked the article. TBZ modified and polished the article, and reviewed the article. All authors contributed to the article and approved the submitted version.
FundingThis review was supported by provincial science and technology innovation strategy special project funding program (Shankefu[2021]88-28).
AcknowledgmentsFigures are created with BioRender.com.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
2. Tian J, Zhang D, Yao X, Huang Y, Lu Q. Global epidemiology of systemic lupus erythematosus: a comprehensive systematic analysis and modelling study. Ann Rheum Dis (2023) 82:351–6. doi: 10.1136/ard-2022-223035
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med (2017) 23:615–35. doi: 10.1016/j.molmed.2017.05.006
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Yap DYH, Chan TM. B cell abnormalities in systemic lupus erythematosus and lupus nephritis-role in pathogenesis and effect of immunosuppressive treatments. Int J Mol Sci (2019) 20:6231. doi: 10.3390/ijms20246231
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Li H, Boulougoura A, Endo Y, Tsokos GC. Abnormalities of T cells in systemic lupus erythematosus: new insights in pathogenesis and therapeutic strategies. J Autoimmun (2022) 132:102870. doi: 10.1016/j.jaut.2022.102870
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol (2008) 181:8761–6. doi: 10.4049/jimmunol.181.12.8761
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Alexander JJ, Jacob A, Chang A, Quigg RJ, Jarvis JN. Double negative T cells, a potential biomarker for systemic lupus erythematosus. Precis Clin Med (2020) 3:34–43. doi: 10.1093/pcmedi/pbaa001
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Liu J, Zhang X, Cao X. Dendritic cells in systemic lupus erythematosus: from pathogenesis to therapeutic applications. J Autoimmun (2022) 132:102856. doi: 10.1016/j.jaut.2022.102856
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Rekvig OP, van der Vlag J. The pathogenesis and diagnosis of systemic lupus erythematosus: still not resolved. Semin Immunopathol (2014) 36:301–11. doi: 10.1007/s00281-014-0428-6
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Bolouri N, Akhtari M, Farhadi E, Mansouri R, Faezi ST, Jamshidi A, et al. Role of the innate and adaptive immune responses in the pathogenesis of systemic lupus erythematosus. Inflammation Res (2022) 71:537–54. doi: 10.1007/s00011-022-01554-6
CrossRef Full Text | Google Scholar
14. Woo JMP, Parks CG, Jacobsen S, Costenbader KH, Bernatsky S. The role of environmental exposures and gene-environment interactions in the etiology of systemic lupus erythematous. J Intern Med (2022) 291:755–78. doi: 10.1111/joim.13448
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Teruel M, Alarcón-Riquelme ME. The genetic basis of systemic lupus erythematosus: what are the risk factors and what have we learned. J Autoimmun (2016) 74:161–75. doi: 10.1016/j.jaut.2016.08.001
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Wolf SJ, Estadt SN, Gudjonsson JE, Kahlenberg JM. Human and murine evidence for mechanisms driving autoimmune photosensitivity. Front Immunol (2018) 9:2430. doi: 10.3389/fimmu.2018.02430
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Gonzalez-Quintial R, Mayeux JM, Kono DH, Theofilopoulos AN, Pollard KM, Baccala R. Silica exposure and chronic virus infection synergistically promote lupus-like systemic autoimmunity in mice with low genetic predisposition. Clin Immunol (2019) 205:75–82. doi: 10.1016/j.clim.2019.06.003
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Illescas-Montes R, Corona-Castro CC, Melguizo-Rodríguez L, Ruiz C, Costela-Ruiz VJ. Infectious processes and systemic lupus erythematosus. Immunology (2019) 158:153–60. doi: 10.1111/imm.13103
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Kim JW, Kim HA, Suh CH, Jung JY. Sex hormones affect the pathogenesis and clinical characteristics of systemic lupus erythematosus. Front Med (Lausanne) (2022) 9:906475. doi: 10.3389/fmed.2022.906475
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients (2012) 4:1095–119. doi: 10.3390/nu4081095
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Stanisavljević S, Lukić J, Momčilović M, Miljković M, Jevtić B, Kojić M, et al. Gut-associated lymphoid tissue, gut microbes and susceptibility to experimental autoimmune encephalomyelitis. Benef Microbes (2016) 7:363–73. doi: 10.3920/BM2015.0159
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Chen BD, Jia XM, Xu JY, Zhao LD, Ji JY, Wu BX, et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol (2021) 73:232–43. doi: 10.1002/art.41511
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Huang C, Yi P, Zhu M, Zhou W, Zhang B, Yi X, et al. Safety and efficacy of fecal microbiota transplantation for treatment of systemic lupus erythematosus: an EXPLORER trial. J Autoimmun (2022) 130:102844. doi: 10.1016/j.jaut.2022.102844
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol (2018) 84:e02288-17. doi: 10.1128/AEM.02288-17
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Zegarra-Ruiz DF, El Beidaq A, Iñiguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, et al. A diet-sensitive commensal lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe (2019) 25:113–27.e6. doi: 10.1016/j.chom.2018.11.009
留言 (0)