
記住我
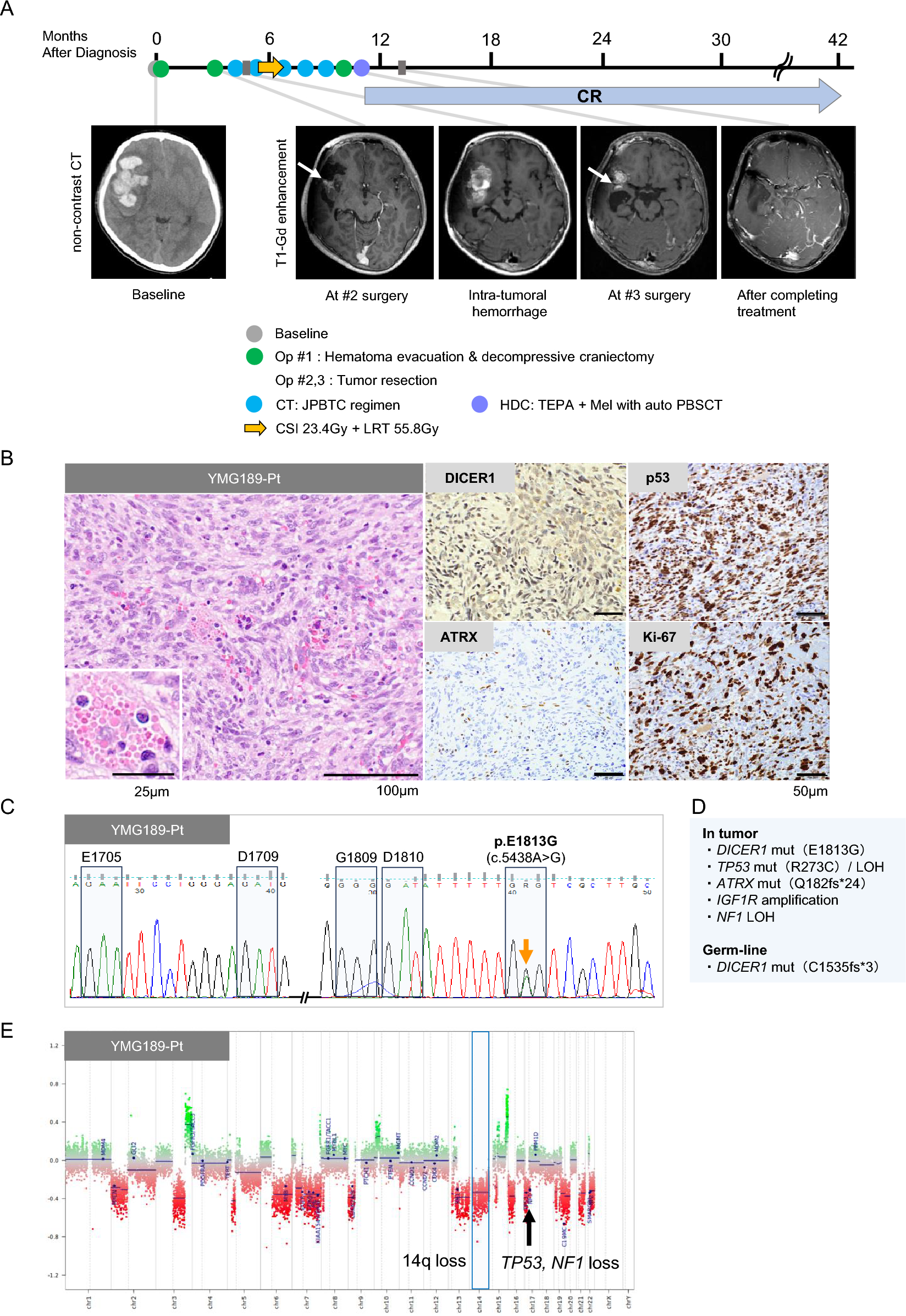





A 72-year-old male patient presented to a local hospital after experiencing diplopia for 1 month. He had had a malignant fibrous histiocytoma of the right thigh and prostate cancer when he was in his 40 s and 60 s, respectively. He also had a significant family history of cancer: his son, sister, and brother had colon cancer, his mother had rectal cancer, his maternal aunt had stomach cancer, and his maternal uncle had liver cancer (Fig. 1).
Fig. 1
Patient’s pedigree showing a family history of cancer and the patient’s history of multiple cancers. The arrow indicates the presenting patient. Filled circles or squares indicate individuals who developed cancer. Ca. cancer, MFH malignant fibrous histiocytoma, y year-old, d. dead
Magnetic resonance imaging revealed a tumor extending from the clivus into the sphenoid sinus. The preoperative diagnosis was clival chordoma (Fig. 2a), and gross total resection was performed using an endoscopic endonasal transsphenoidal approach (Fig. 2b). The histopathological diagnosis was conventional chordoma. However, 4 months postoperatively, the patient experienced diplopia and left facial pain. A 3 cm in diameter recurrent lesion was observed (Fig. 2c), and the lesion shrank after the patient underwent Gamma Knife irradiation. However, 3 months after irradiation, recurrence was observed outside the irradiated area (Fig. 2d), and the patient was re-irradiated. Nine months after the first irradiation, the tumor recurred in the left cavernous sinus (Fig. 2e), and the patient was irradiated for a third time. However, the tumor continued to grow, extending toward the maxillary sinus (Fig. 2f). Subsequent genetic analysis of the chordoma revealed a high-TMB tumor, and ICI pembrolizumab, a humanized anti-programmed cell death protein 1 (PD-1) monoclonal antibody, was indicated. Conversely, before treatment could be initiated, the patient had hematuria and cancer of the renal pelvis. The recurrent chordoma continued growing, and his symptoms, including left facial pain, worsened. 22 months postoperatively (18 months after the first irradiation; Fig. 2g), the first pembrolizumab dose was administered, and the patient underwent a right nephrectomy for cancer of the renal pelvis, and the histopathological diagnosis was papillary urothelial carcinoma. The tumor grew during the period that the first two doses of pembrolizumab were administered (Fig. 2h). However, after the third dose, the tumors began to shrink. Notably, the patient’s symptoms improved considerably after the seventh dose of pembrolizumab (Fig. 2i). The patient continued to receive treatment with pembrolizumab; his left facial pain is currently considerably reduced, and the left abducens palsy tends to improve.
Fig. 2
T2-weighted magnetic resonance images of the tumor at different stages of its development. a preoperatively; b 1 month postoperatively; c at first recurrence, 4 months postoperatively; d at second recurrence, 3 months after irradiation. The tumor has recurred outside the irradiated area; e 9 months after the first irradiation. The tumor has extended into the left cavernous sinus; f the tumor extends toward the maxillary sinus 16 months after initial irradiation; g 18 months after the first irradiation, the tumor continues to grow; h after two doses of an immune checkpoint inhibitor (pembrolizumab), the tumor is still growing; and i after the third dose of pembrolizumab, the tumor begins to shrink. It continues to shrink for the remainder of the course of treatment (seven doses in total)
Pathological findingsThe pathological diagnosis was performed according to the World Health Organization Classification of Tumors, Soft Tissue, and Bone Tumors (5th ed.). The tumor comprised epithelioid cells with lightly eosinophilic cytoplasm, forming short chords, nests, and single cells within the extracellular myxoid matrix. Physaliphorous cells with vacuolated bubbly cytoplasm were also present. Larger epithelioid cells with clear to light eosinophilic cytoplasm were arranged in a solid sheet-like pattern, with minimal to a completely absent extra myxoid matrix. Nuclear atypia and pleomorphism were heterogeneous throughout the neoplasm, ranging from mild to relatively severe. Multinucleated cells with bizarre nuclei were occasionally observed. There were two mitotic figures/10 HPF (field of view diameter: 0.55 mm), both on hematoxylin and eosin- and phospho-histone H3-stained section. The Ki-67 labeling index was 2–3%. Necrosis was not evident. Immunohistochemically, tumor cells were positive for brachyury. There was no complete loss of INI1 (SMARCB1) or BRG1 (SMARCA4) expression. Based on these findings, this tumor was diagnosed as a conventional chordoma (Fig. 3). Interestingly, multiple scattered foci of lymphocytes in the intratumoral region were present. Most intratumoral lymphocytes were positive for PD-1. Approximately 1–2% of tumor cells showed programmed death-1 (PD-L1) partial or complete membrane staining at weak to moderate intensity. PD-L1 displayed weak cytoplasmic staining only in < 1% of immune cells, including lymphocytes.
Fig. 3
Histopathology. H&E staining at a 100x, b 400x, and c 400x. Findings of the immunohistochemical staining (all 200x) for d brachyury, e INI1, f BRG1, g Ki-67, h PD-1, and i PD-L1. Typical histological features of conventional chordoma with abundant extracellular matrix a, b the area is composed of cohesive sheets of larger epithelioid cells with relatively severe nuclear atypia and pleomorphism. No myxoid stroma is present (c). Diffuse nuclear staining for brachyury (d). Intact INI1 (e) and BRG1 (f) expression. PD-1-positive intratumoral lymphocytes (h). PD-L1-positive tumor cells (i). H&E hematoxylin and eosin; PD-L1 programmed death-ligand 1
The primary antibodies used were as follows: INI1 (SMARCB1) (clone: 3E10, abnova, Taipei, Taiwan) at dilution of 1:200, BRG1 (SMARCA4) (clone: EPR 3912, Abcam, Cambridge, UK) at dilution of 1:100, PD-1 (clone: NAT105, abcam, Cambridge, UK) at dilution of 1:50, and PD-L1 (clone SP142, Spring Bioscience, Pleasanton, CA, USA) at dilution of 1:500.
Genetic analysis of the chordomaGenome profiling using FoundationOne® CDx (Foundation Medicine, Inc., Cambridge, MA) was performed using genomic DNA extracted from formalin-fixed paraffin-embedded tissue of the tumor specimen. The results were equivocal regarding microsatellite instability; nonetheless, the TMB was high at 10 mutations per megabase (Muts/Mb), and mutations were also identified in the APC (L616fs*18), CD79B (Y196H), INPP4B (splice site 2375-2A > G), JAK1 (K860fs*16), MSH6 (F1088fs*5), and MLH1 (S698fs*5) genes. The genome panel test revealed mutations in the following two DNA mismatch repair (MMR) genes observed in Lynch syndrome: MLH1 and MSH6.
Diagnosis of Lynch syndromeGermline genetic testing for MLH1 and MSH6 was undertaken using genomic DNA extracted from the patient’s blood sample, and a variant was identified in MLH1, c.2092_2093del (p.Ser698Argfs*5). No pathological variants were detected for MSH6, a somatic variant. This, together with the history of cancer in the patient and his family, as shown in Fig. 1, led to a diagnosis of Lynch syndrome.
留言 (0)