In this study, we conducted a retrospective review about the clinical, radiological, pathological, and molecular characteristics of TG cases in our institute. Although the clinical course of TG which is mostly diagnosed as PA or LrGG is typically indolent, the median PFS and OS were 3.4 years and 6.2 years, respectively, in our series. Previous reports showed that the clinical course of typical PA after a gross total resection is usually favorable, with a 10-year overall survival (OS) rate of approximately 95% [2, 15, 26, 28]. However, Liu et al. [12] reported that 121 of 453 (26.7%) TG patients experienced clinical and/or radiological progression, with the mean time from diagnosis to progression ranging from 3 months to 7.8 years. Thus, these findings suggested that TG may exhibit relatively poorer PFS and OS compared to typical PA or LrGGs in other locations and TG is not always an indolent tumor.
Regarding the pathological analysis, our study identified that TG included diverse diagnosis such as pilocytic astrocytoma, glioblastoma, diffuse high-grade glioma, and DMGs H3 K27-altered. Previously the histopathological findings of TG are typically associated with LrGG such as PA or diffuse astrocytoma. In a systematic review of 355 pediatric TG patients, Bauman et al. [1] reported that, among 117 pathologically classifiable cases, 37.6% had PA, 25.6% had diffuse astrocytoma, and 12.8% had low-grade (non-pilocytic) glioma. In contrast, Mohme et al. [19] reported that in 12 of 23 pathologically classifiable cases, 58.3% were LrGG (5 PA, 1 ganglioglioma, and 1 diffuse astrocytoma), while 42.7% were classified as HGGs (3 glioblastomas, and 2 anaplastic astrocytomas). Other reported pathological findings of TG include morphologically PA with genetic H3 K27M mutation, pleomorphic xanthoastrocytoma, and atypical teratoid/rhabdoid tumors [20, 21, 25, 27, 30]. These findings suggest that TG has heterogeneous pathology, despite being predominantly an LrGG.
Regarding the molecular analysis, the KRAS mutations and BRAF alterations have recently been observed in TG. In addition, the genome-wide DNA methylation profile indicates that TG is a distinct tumor in comparison to other LrGGs [6, 8, 11, 12]. Chiang et al. [6] reported a high frequency of KRAS G12R mutations (82.6%) and BRAF mutations (60.9%) in their analysis of 23 TG cases. In addition, other reported mutations in TG include KRAS p.E63K missense mutation and KRAS c181C>A mutation [5, 31, 32]. Liu et al. [12] conducted genome-wide DNA methylation analysis in 45 cases of TG, revealing a distinct methylation pattern compared to PAs at other sites. In our study, two patients (Case 2 and 6) underwent F1CDx, and both harbored KRAS alterations (KRAS G12A and KRAS G13R in Case 2, and KRAS Q61R in Case 3), which suggested that there may be a class of TGs with KRAS mutation. The molecularly evaluated TG reports from the last decade are summarized in Table 2. KRAS mutations and BRAF alterations have been reported relatively frequently in TG with LrGG such as PA and DA, while a few mutations like H3 K27M and SMARCB1 have been observed less frequently. This study also identified KRAS and H3 K27M mutations in two cases histologically confirmed with LrGG (Table 2).
Table 2 Summary of literature on tectal glioma with molecular analysisAlthough RAS mutations are generally rare in gliomas and not associated with a specific tissue phenotype [17], a relatively large proportion of TGs have been reported KRAS mutations and BRAF alterations. KRAS is involved in several signaling cascades, including MAPK, PI3K/ACT, RAL-GDS, JAK/STAT3, and NORE1/RASSF1 pathways, influencing various cellular processes such as growth, apoptosis, growth arrest, differentiation, transcription, and translation [5, 17, 22, 31]. For example, KRAS signaling is essential for the maintenance of glioblastoma growth maintenance in mice, suggesting that inhibition of KRAS leads to tumor apoptosis [3, 7]. In addition to hotspot mutations in KRAS, Yakir et al. [32] reported SRGAP3::RAF1 fusion in pediatric TG. SRGAP3::RAF1 fusion has also been reported in pediatric PA cases and abnormally activates both MAPK and PI3K/mTOR signaling pathways [9, 23]. These findings support that there is a group of TGs with mutations in KRAS or its downstream pathway, exhibiting a distinct methylation pattern and differing entity from other LrGGs. However, the detailed molecular landscape of KRAS alterations in TG remains unclear, and further analysis is required.
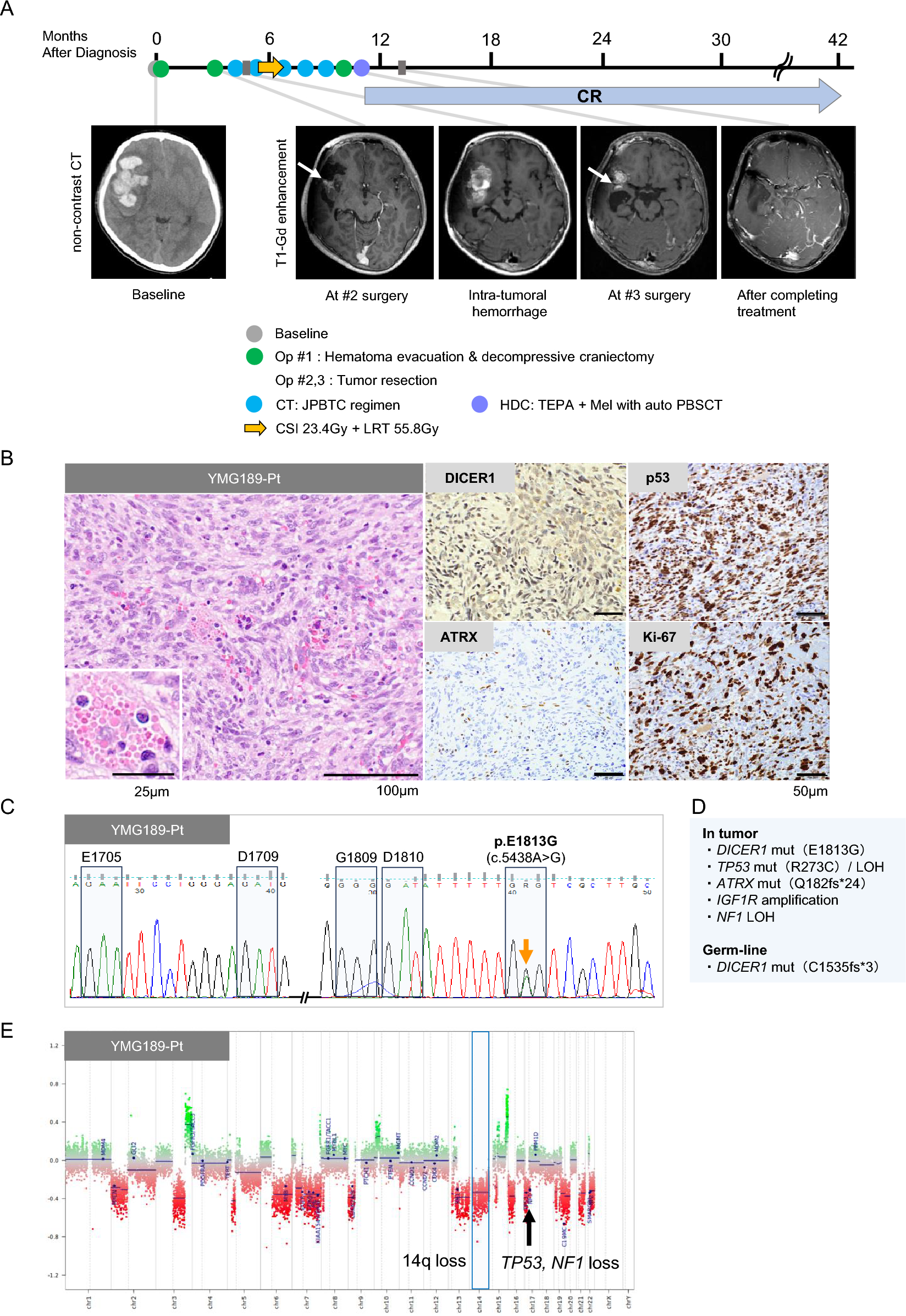
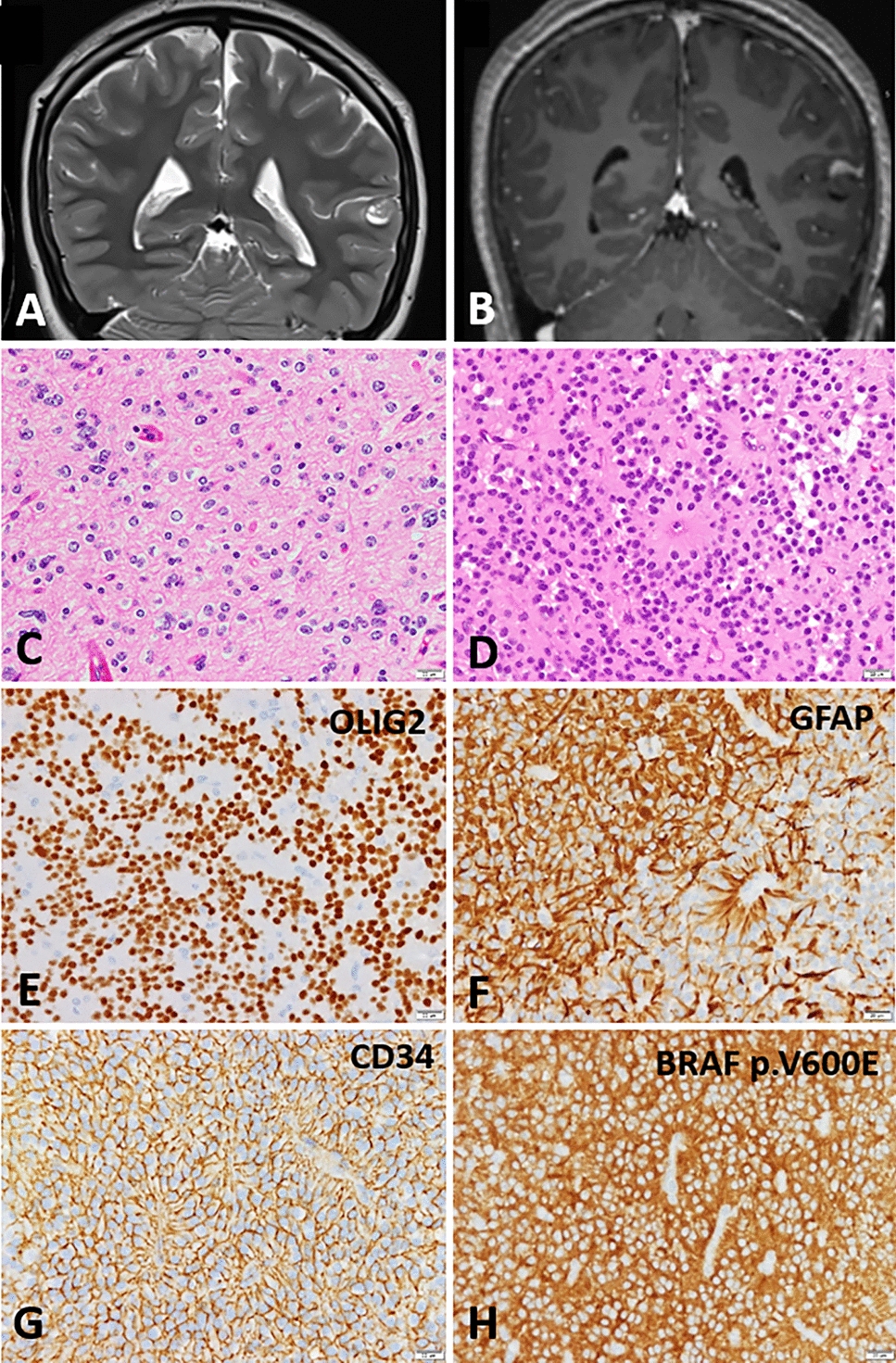
In our study, Case 3 was pathologically diagnosed as PA, and H3 K27M mutation was inferred by genome-wide DNA methylation profiling and immunohistochemistry (Fig. 3D–F). We have identified three reports of TG with histologic findings of PA and H3 K27M mutation, including our case [20, 25]. In the 2021 WHO Classification of Tumors of the Central Nervous System, DMGs were renamed DMGs H3 K27-altered, considering the various mechanisms affecting the epigenetic pathway [13, 14, 24]. Conversely, several non-DMG tumors, such as ependymoma, PA, pediatric diffuse astrocytoma, and ganglioglioma, have been reported to harbor the same H3 K27M mutation, displaying a different clinical presentation and a favorable prognosis compared to typical DMGs H3 K27-altered [13, 20, 25]. Sumi et al. described these as nontypical DMGs. Two reported cases of morphologic PA with H3 K27M mutation showed no recurrence during observation periods of 8 and 28 months, respectively (Table 2). Case 3 had a PFS of 4.5 years and an OS of 7.2 years (Table 1). These results suggest that PAs with H3 K27M mutations tend to have a relatively better prognosis compared to typical DMG H3 K27-altered patients. We consider the PA with H3 K27M mutation in our study to be a part of this group, often referred to as nontypical DMGs.
We summarized previous reports of TGs that have been evaluated molecularly in Table 2. Given the diverse histological and molecular features of TG, there is a potential discrepancy between histological and molecular evaluations. Thus, TG requires detailed histological and molecular analysis including genome-wide DNA methylation, and further accumulation of knowledge is needed to establish this pathogenesis as a clear entity.
Limitations of this study include its retrospective design, small number of cases, and the lack of genetic information in some cases. In addition, although we included only symptomatic cases in this study, evaluating asymptomatic cases with an untreated imaging course is considered necessary.






留言 (0)