
記住我
Obesity is a rapidly growing danger that has reached worldwide epidemic proportions, with half of the US population predicted to be obese by 2030 (Wang et al., 2011). Obesity is an independent risk factor and a comorbid condition to many diseases, including atherosclerotic cardiovascular disease (Rocha and Libby, 2009; Powell-Wiley et al., 2021), diabetes (Barnes, 2011), steatohepatitis (Li et al., 2016) and neurodegenerative diseases (Pugazhenthi et al., 2017). All these diseases are characterized by chronic inflammation involving macrophages as mediators of inflammation. There are multiple overlaps in the pathophysiology of these conditions, including infiltrating monocytes and their differentiation into macrophages, subsequently resulting in uptake of excess lipids, followed by cell retention and death. Inflammation is at the heart of obesity-related conditions and macrophages have a complicated role in both the promotion and alleviation of this inflammation (Figure 1).
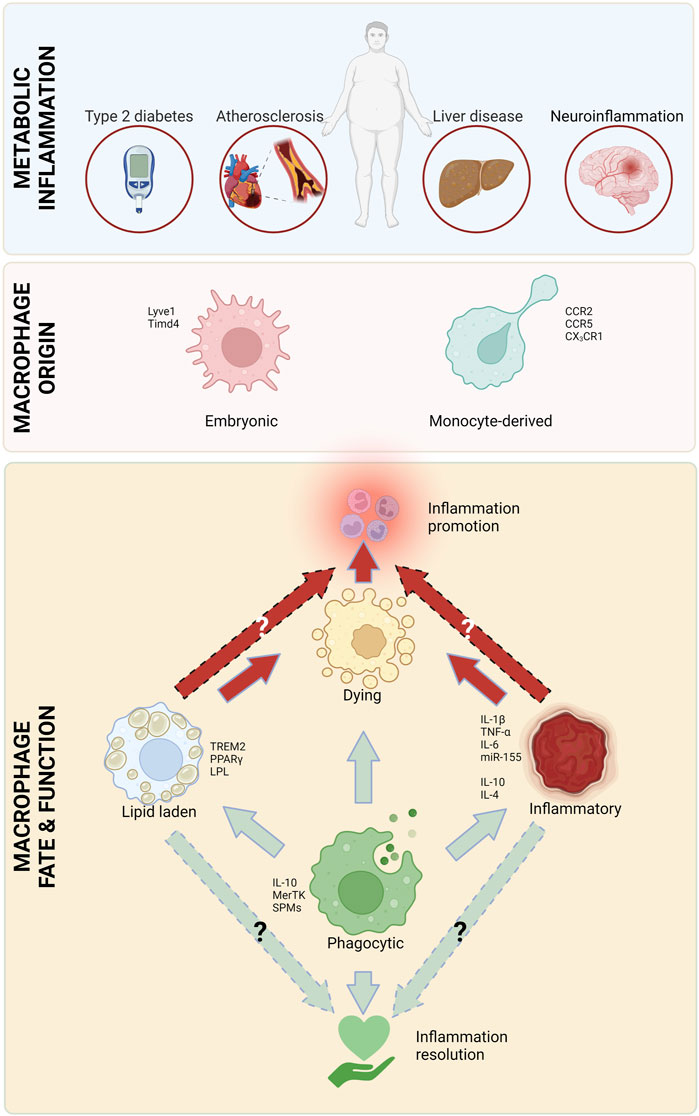
FIGURE 1. Major macrophage attributes in metabolic inflammation. Macrophages, originating from circulating monocytes and embryonic tissues play a critical role in all types of metabolic inflammation. There are several types of macrophages that are involved in inflammation initiation and heightening (red arrows), as well as resolution (green arrows). Created with Biorender.com.
Our knowledge about macrophages has evolved from them being immunological first responders to being integral players in tissue homeostasis, and major regulators of systemic and local tissue metabolism. In this way, macrophages play a critical role in balancing the immune response and ensuring that inflammation is resolved in a timely and appropriate manner. Here, we highlight the most important functions of macrophages, including phagocytosis, lipid uptake and excretion, and the production of multitude of inflammation regulating mediators, and how they contribute to the pathogenesis and resolution of metabolic diseases.
Macrophage originMacrophages can be divided into two main types based on their origin: resident and newly recruited (monocyte-derived). Both types of macrophages play important, yet distinct roles in the immune system and the maintenance of tissue health. Resident macrophages are permanently present in tissues. These cells are derived from embryonic tissues (yolk sac and the aorta-gonad-mesonephros-fetal liver axis (Yona et al., 2013; Gomez Perdiguero et al., 2015; Hoeffel et al., 2015; Wu and Hirschi, 2020)) and remain in the tissue throughout the lifetime of the organism. Resident macrophages are important for maintaining the normal tissue function and can also respond rapidly to tissue damage. Tissue macrophages are often maintained by local proliferation, although in some tissues their maintenance requires replenishment from circulating monocytes [reviewed in (Gordon and Pluddemann, 2017)]. Monocyte-derived macrophages, on the other hand, are produced from circulating monocytes that can migrate to tissues when needed. In some situations, monocytes can assume a tissue-resident macrophage phenotype (Zhao et al., 2018). Once in the tissue, these cells differentiate into macrophages and play a key role in the immune response. Monocyte-derived macrophages can be found in almost all tissues in the body and are particularly important for responding to infections and other types of tissue damage. Macrophage numbers in tissues are regulated by dynamic processes, such as recruitment (Swirski et al., 2007; Tacke et al., 2007; Rahman et al., 2017), proliferation (Robbins et al., 2013), retention/stasis (van Gils et al., 2012; Wanschel et al., 2013; Ramkhelawon et al., 2014; Aziz et al., 2017), egress (Bradfield et al., 2016) and apoptosis (Linton et al., 2016) (reviewed in (Weinstock and Fisher, 2019)). In this review we will mainly focus on the differences between embryonic and monocyte-derived macrophages, and will, thus, mostly mention aspects of proliferation/survival and recruitment.
Lipid handlingMacrophages can take up and break down various types of lipids and lipid particles, including low-density lipoprotein (LDL), high-density lipoprotein (HDL), very low-density lipoprotein (VLDL), which mainly carry triglycerides and cholesterol. Once lipid particles are engulfed, they are broken down, and the resulting products are either used by the macrophage for energy or are further processed and released back into the bloodstream or tissue (Remmerie and Scott, 2018). During inflammation, macrophages typically retain lipids in intracellular lipid droplets, giving these cells the microscopic appearance of containing foam, and were thus termed “foam cells”. Lipid droplets are organelles that store and control the release of lipids. They also regulate inflammation by providing the energy to support several immune processes, such as microbial clearance, production of inflammatory mediators and antigen presentation [reviewed in (den Brok et al., 2018)]. The accumulation of excess fat is a result of multiple factors including dysregulation of influx-efflux pathways and fatty acid oxidation. Importantly, macrophages can clear excess lipids, such as cholesterol, and direct their delivery to the liver for redistribution, breakdown or excretion (Tall and Yvan-Charvet, 2015).
CytokinesDuring the inflammatory response, cytokines are released by immune and non-immune cells and help to recruit other immune cells to the site of injury or infection. Furthermore, they play a key role in regulating the intensity and duration of the inflammatory response. Some of the key cytokines that are involved in promoting metabolic inflammation include interleukin (IL)-1, IL-6, tumor necrosis factor-alpha (TNFα), and interferon (IFN) ɣ (Fujiwara and Kobayashi, 2005). Cytokines such as IL-10 and IL-4 also help to fine-tune the specific immune response needed for inflammation resolution and mediate the repair of tissue damage, to prevent chronic inflammation which can lead to long term tissue damage (Watanabe et al., 2019). Overall, the balance between pro- and anti-inflammatory cytokines is essential for the proper functioning of the immune system.
Lipid mediatorsOmega-3 and omega-6 are a large family of polyunsaturated fatty acids (PUFAs) found in a variety of foods, including fish (mostly omega-3 rich), nuts, seeds, and vegetable oils (mostly omega-6 rich), and are essential for good health. PUFAs have been shown to have anti-inflammatory effects, presumably by inhibiting the production of certain inflammatory molecules, such as thromboxanes and leukotrienes. Furthermore, PUFAs may help to reduce the activity of certain immune cells, such as T and B cells (reviewed in (Calder, 2006)]. Although there is conflicting evidence for the efficacy of either (Ramsden et al., 2013; Ramsden et al., 2016; Abdelhamid et al., 2018), omega-3 is generally considered to have more beneficial effects on health than omega-6 PUFAs, since they are the precursor for many pro-resolving mediators. The most investigated omega-3 fatty acids are eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA).
REDUCE-IT is a large, randomized controlled trial that evaluated the effectiveness of icosapent ethyl, a form of EPA, in reducing the risk of cardiovascular events in high-risk patients. The results show that treatment with EPA reduces the risk of major cardiovascular events by 25% compared to placebo (Bhatt et al., 2019). This is the first major evidence in humans that lipid mediators may present an effective therapy for metabolic diseases. However, this is an ongoing line of investigation, as a subsequent large clinical trial showed no benefit of a combined formulation of EPA and DHA on major cardiovascular events (Nicholls et al., 2020).
Alongside PUFAs, other lipid mediators have been found to modulate inflammation. Arachidonic acid (which can be derived from PUFAs) is the precursor of several mediators implicated in both inflammation promotion and resolution. In macrophages and other cells, arachidonic acid can be converted to pro-inflammatory leukotrienes (LT), such as LTB4, or to pro-resolving mediators, such as lipoxin A4 (LXA4) or DHA and resolvins (reviewed in) (Basil and Levy, 2016). Arachidonic acid can also be metabolized by cyclooxygenase (COX; gene name Prostaglandin-endoperoxide synthase, Ptgs)-1/2 to prostaglandins (PGs), which were shown to have both pro- and anti-inflammatory properties, according to their location, concentration and cellular target (Vane et al., 1998).
Specialized Pro-resolving Mediators (SPMs) are a class of lipid-derived effectors that are biosynthesized at the site of inflammation and work by activating various G-protein-coupled receptors. They play a critical role in balancing the pro-inflammatory mediators and have been implicated in promoting efferocytosis (Basil and Levy, 2016). SPMs are broadly categorized into lipoxins, resolvins (Rv), protectins and maresins (MaR). They have been found to be protective in vivo (in several rodent species), where they promote the clearance of microorganisms, debris and apoptotic cells, and promote tissue repair independently of anti-inflammatory signals (Basil and Levy, 2016).
MicroRNAs (miR)miRs are small non-coding RNAs that play a key role in regulating gene expression. They do this by binding to specific target genes and either inhibiting their transcription, translation or influencing their mRNA stability. miRs direct multiple processes by regulating the expression of genes in macrophages which play an important role in maintaining tissue integrity (Liu and Abraham, 2013). Interestingly, miRs are also a mean of cell-cell communication, as they can be transferred through exosomes (Hu et al., 2012) and lipoproteins (Michell and Vickers, 2016).
Efferocytosis and phagocytosisBillions of cells naturally die every day in healthy individuals. These cells are being phagocytosed and cleared by macrophages in a process termed efferocytosis. Since inflammation drives cell death, which in turn exacerbates inflammation, efferocytosis is critical for inflammation resolution. Macrophages recognize apoptotic cells via ligand-receptor interactions, followed by engulfment of the dead cells or debris [reviewed in (Thorp and Tabas, 2009)]. This induces phagosome-lysosome fusion and degradation of the engulfed contents. A defective efferocytosis pathway is one of the hallmarks of chronic inflammatory diseases, resulting in accumulation of dead cells, which enhances inflammation and creates a feed-forward loop of inflammation and cell death. Efferocytosis is a quieter form of phagocytosis; while phagocytosis is followed by antigen presentation and T cell activation, in efferocytosis, internalization of apoptotic cells leads to its sequestration towards endosomes, taking it away from MHCII- mediated antigen presentation (Yin and Heit, 2021).
Adipose tissue inflammation and insulin resistanceDiabetes, one of the major public health challenges worldwide, is estimated to be affecting 537 million people between ages 20–79. This number is estimated to increase to 784 million by 2045 (Federation, 2021). The increase in the prevalence of type 2 diabetes is not surprising, since obesity is one of its major causes (Dale et al., 2017). Apart from the economic burden posed by diabetes, it is also a major driver of mortality. In 2021 alone, ∼6.7 million adults died as a result of diabetes complications, without accounting for COVID-19-related mortality that is exacerbated by diabetes (Federation, 2021).
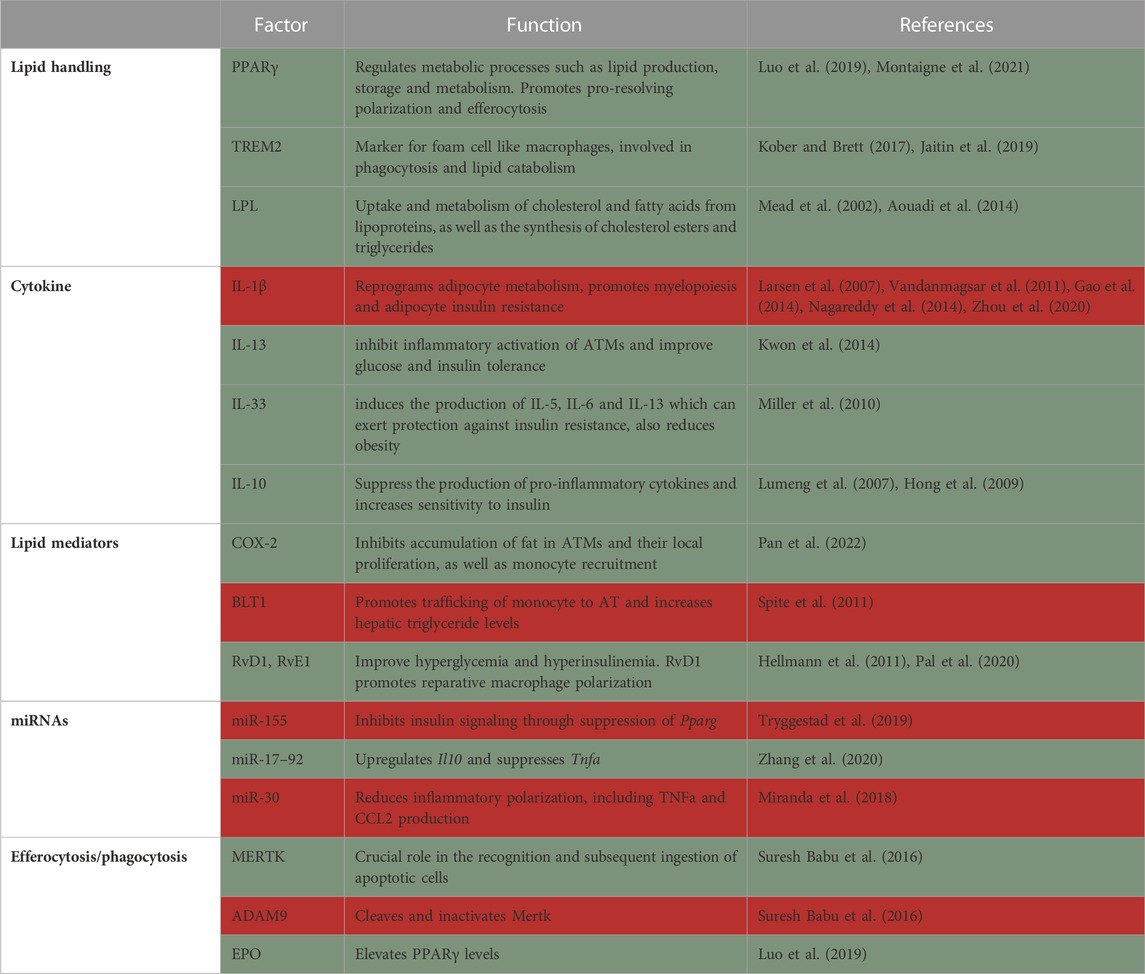
The main role of the adipose tissue (AT) is to store excess nutrients, mostly in the form of triacylglycerols, and release these lipids in times of energetic needs, e.g., exercise, under-nutrition etc. [reviewed in (Heffron et al., 2020)]. Obesity induces extensive remodeling of the AT, including tissue expansion and changes in structure and cellular composition. AT expansion is caused by adipocyte hyperplasia (cell number increase) and hypertrophy (cell enlargement) to allow the storage of excess lipids (Berry et al., 2014). The inability to appropriately expand AT in obesity leads to ectopic lipid deposition in the liver and skeletal muscle and may be an underlying cause of insulin resistance. These processes also cause dramatic changes to the immune milieu in the adipose (Jaitin et al., 2019; Weinstock et al., 2019). Macrophages play a critical role in lipid storage and utilization in the AT, tightly regulating metabolic health [reviewed in (Peterson et al., 2018)]. There is an active debate regarding the role of AT macrophages (ATMs), with early studies suggesting they promote metabolic pathologies such as insulin resistance. However, the common modern view is that ATMs are multifunctional, with some having detrimental and some beneficial effects on metabolic inflammation (Table 1) (Coats et al., 2017; Weinstock et al., 2020).
TABLE 1. Summary of factors in macrophages that protect from (green) or promote (red) adipose tissue inflammation and insulin resistance.
Macrophage originSeveral studies have investigated the origin of AT macrophages in obesity. In a seminal study, Weisberg et al. found that ATMs are derived from bone marrow progenitors (Weisberg et al., 2003). By performing bone marrow transplantation, they showed that 85% of ATMs were derived from the donor bone marrow 6 weeks post-transplantation. This ATM accumulation preceded the development of insulin resistance, and was proposed to be the causal link between obesity and insulin resistance (Xu et al., 2003).
Two studies discovered that increased levels of chemokine (C-C motif) ligand 2 (CCL2) in AT and plasma is responsible for increased macrophage accumulation in obesity, thus suggesting monocyte recruitment to the AT. They found that there was upregulation of CCL2 in the adipose tissue and plasma of genetically obese (db/db) and high-fat diet (HFD)-fed mice. In both studies, a transgenic mouse overexpressing CCL2 was created, which was sufficient to induce macrophage infiltration into AT, insulin resistance, and increase hepatic triglyceride content, even with low-fat diet feeding (Kamei et al., 2006; Kanda et al., 2006). On the other hand, CCL2 global deficiency reduced ATM accumulation and improved insulin intolerance and hepatic steatosis associated with adiposity in diet-induced obese (DIO) mice (Kanda et al., 2006). Along the same lines, C-C chemokine receptor type 2 (CCR2), the receptor for CCL2, CCL7 and CCL8, is also involved in the development of metabolic dysfunction and ATM recruitment. In fact, obesity causes dramatic upregulation of CCL2, CCL7 and CCL8 in the AT (Weisberg et al., 2006). Knockout of Ccr2 in DIO mice shows lower fasting blood glucose and improved glucose and insulin tolerance (Weisberg et al., 2006). Subsequently, others have also showed the importance of CCR2 in the development of metabolic dysfunction and ATM accumulation in obese mice (Ito et al., 2008; Sullivan et al., 2013). Contrarily, a recent study found that resident ATMs do not express CCR2, but rather Timd4, derived from early embryonic tissues. These resident ATMs produce platelet-derived growth factor (PDGF) cc that regulates lipid storage in adipocytes. HFD increases Pdgfc expression in ATMs, and its blockade prevents adipocyte hypertrophy, possibly by downregulating adipogenesis (Cox et al., 2021). Further studies, using cell depletion, show that resident ATMs are integral to metabolic health (Chen et al., 2021). CD169, which is presumably expressed more on non-recruited ATMs was used to deplete this population, without affecting monocyte-derived ATMs. Absence of CD169+ macrophages increased AT mass following HFD and promoted adipocyte hypertrophy. CD169+ ATMs were proposed to maintain the AT vasculature integrity (Chen et al., 2021). An earlier study elegantly described ATMs that are in close contact with vasculature and were termed vascular-associated macrophages (Silva et al., 2019). These cells were shown to protect the AT with their high endocytic capacity, thus creating a buffer from the circulation and preventing the entrance of insults to the AT. Obesity impairs the endocytosis ability of vascular-associated macrophages, allowing more promiscuous entry of substances from the blood to the AT (Silva et al., 2019).
HFD does not only increase total ATM numbers but also attracts a unique ATM subpopulation that is minimally present in AT under steady state conditions. The obesity-driven infiltrating ATMs are bone marrow-derived, become lipid-laden (foam cells) and express markers such as Cd11c, Cd9, and Trem2. The majority of these infiltrating ATMs form classical crown-like structures around dead or damaged hypertrophic adipocytes. CD11c+ ATMs in obese mice were shown to possess high lipid metabolism and bioenergetic activity. CD11c+ ATMs are equipped with specific receptors (e.g., Cd36, Msr1) and enzymes (e.g., Lpl, Fabp4, and Mgl1) to take up and process lipids. Altogether it seems that monocyte-derived and resident ATMs have distinct roles in regulating AT functions, which requires further elucidation.
Lipid handlingLipid handling is important in macrophages to maintain metabolic health. This was suggested in an early study that investigated the role of PPAR-γ in macrophages. PPAR-γ is a transcription factor and master regulator of metabolic processes, including lipid production, storage and metabolism [reviewed in (Montaigne et al., 2021)]. Macrophage-specific knockout of Pparg results in glucose intolerance systemically, as well as in the liver and muscle, even in lean conditions (Hevener et al., 2007). In the AT, macrophage Pparg knockout impaired the expression of numerous genes involved in lipid uptake, synthesis and β–oxidation. Furthermore, adipocytes co-cultured with Pparg−/− ATMs show inhibition of insulin-stimulated glucose uptake, directly showing causality of macrophage Pparg deficiency and adipose insulin resistance (Odegaard et al., 2007). Pparg deficiency further prevents the beneficial effects of the anti-diabetic drug Rosiglitazone in obese mice (Hevener et al., 2007).
Trem2 is a hallmark gene for macrophages that accumulate in adipose and other lipid-rich tissues (Kober and Brett, 2017). It is a receptor for multiple ligands, including lipoproteins and phospholipids, and is a major driver of tissue-level immune cell remodeling [reviewed in (Kober and Brett, 2017)]. Trem2 is expressed on a subset of ATMs, which were termed lipid-associated macrophages (LAM). LAMs arise from circulating monocytes and are positioned around enlarged adipocytes. Functionally, LAMs are thought to be involved in phagocytosis and lipid catabolism, through a TREM2-induced transcriptional program. Consequently, Trem2 deficiency prevents the formation of crown-like structures in the obese AT, causing massive adipocyte hypertrophy, systemic hypercholesterolemia, inflammation, and glucose intolerance (Jaitin et al., 2019). Hence, Trem2 expression limits obesity-induced metabolic dysfunction, possibly by promoting the removal of dysfunctional defective adipocytes by ATMs. Moreover, macrophage accumulation around large adipocytes in obese adipose might be driven by TREM2 to locally contain toxic lipids and other inflammatory mediators from harming their surroundings.
Lipid uptake by ATMs was also shown to modulate systemic glucose tolerance and promote inflammation and metabolic derangement in obesity. For instance, lipoprotein lipase (LPL) is an enzyme that hydrolyzes triglycerides and phospholipids for lipid uptake, which can be used as energy or stored in the form of triglycerides in lipid droplets. LPL is involved in the uptake and degradation of cholesterol and fatty acids from lipoproteins, as well as the synthesis of cholesterol esters and triglycerides (Mead et al., 2002) and its role was extensively investigated in macrophages. In one study, Lpl was knocked down specifically in ATMs using nanoparticles containing siRNA against this gene. Lpl knockdown decreased foam cell formation in visceral AT of obese mice and reduced fatty acid uptake from very low-density lipoprotein (VLDL) hydrolysis. Lpl knockdown in ATMs decreased the expression of genes involved in fatty acid uptake and esterification (Cd36 and Dgat2, respectively), resulting in higher levels of circulating free fatty acids and glucose intolerance in obese mice (Aouadi et al., 2014). That said, more recent studies did not find a role for macrophage LPL in obesity-related morbidity. In this study, the authors compared the effects of genetic global or macrophage-specific deletion of Lpl in obesity, peritonitis and atherosclerosis regression. They demonstrated that ATMs accumulated less fat only in the global, but not macrophage-conditional knockouts, which was associated with a more pro-reparative macrophage phenotype (Chang et al., 2019). In vitro, macrophage Lpl is needed for lipid uptake from VLDL (Chang et al., 2019).
Augmented VLDL-VLDLR signaling was shown in obese ATMs to aggravate AT inflammation and insulin resistance. The presence of VLDLR in macrophages from obese mice increased intracellular level of triglyceride, which stimulates the expression of pro-inflammatory genes, such as Nos2, Tnfa, Ccl2, serum amyloid A, Il1b and Ifng. It was further found that the levels of C16:0 ceramides are decreased in VLDLR deficient macrophages, which are less inflammatory, through the reduced activity of mitogen-activated protein kinases (MAPK) (Kober and Brett, 2017).
ATMs also have an important role in lipid trafficking, independent of their inflammatory phenotype. The expansion of AT during obesity induces a program of lysosome biogenesis in ATMs that is associated with lipid catabolism. This program is induced by factors produced by adipocytes and tightly coupled to lipid accumulation in ATMs. Inhibition of ATM lysosomal function impairs lipid metabolism, increases lipid content in ATMs and reduces AT lipolysis, contributing to worsening of metabolic syndrome (Weisberg et al., 2003). Contrarily, others have shown that lipolysis and lipid storage in macrophages do not affect adipose tissue inflammation. In a recent study, Xanthe et al. demonstrated that hypoxia inducible lipid droplet associated (HILPDA), a protein that promotes lipid storage in macrophages, was dispensable in obesity-induced AT inflammation. They showed that myeloid Hilpda deficiency markedly reduces intracellular lipid levels in macrophages in vitro and in the AT, without affecting inflammation and glucose intolerance (Xu et al., 2003). The discrepancy between these reports might be due to the importance of macrophage lipid uptake and metabolism, rather than storage. Moreover, it is unclear how Hilpda or Lpl deficiency alter the types of lipids utilized by macrophages, which can also greatly influence their phenotype and outcomes of obesity.
CytokinesNumerous classical cytokines play a critical role in adipose tissue inflammation and diabetes. Several seminal papers described the involvement of pro-inflammatory cytokines, namely, IL-1β, in the pathogenesis of obesity and diabetes. Importantly, it was shown in a clinical trial that Anakinra, an IL-1 receptor antagonist, improves systemic inflammation and glycemia, with no effect on insulin sensitivity in type II diabetes patients (Larsen et al., 2007). NLR family pyrin domain containing 3 (NLRP3) is the core component of the inflammasome, responsible for IL-1β processing and secretion. Both Il1b and Nlrp3 gene expression is increased in the obese AT of mice and humans, which is reversible by weight loss (Vandanmagsar et al., 2011). Interestingly, Nlrp3−/− mice fed HFD are more insulin and glucose sensitive, and protected from hepatic steatosis, compared with Nlrp3 sufficient controls (Vandanmagsar et al., 2011). Mechanistically, Nagareddy et al. (2014) showed that ATMs are stimulated by S100A8/A9 that are produced by obese adipocytes. In response, ATMs secrete IL-1β, which activates immune progenitors in the bone marrow to produce more monocytes and neutrophils, thus inducing a feed-forward loop of AT inflammation. IL-1 was further demonstrated to reprogram adipocyte metabolism, exacerbating adipose dysfunction and obesity (Zhou et al., 2020). Others attempted to link insulin resistance itself to macrophage-derived IL-1β. One such study investigated how IL-1β-primed macrophages influence adipocyte insulin signaling in vitro. They found that macrophages treated with IL-1β induce adipocytic expression of pro-inflammatory cytokines (i.e., Ccl2, Il6 and Il8) and blunt insulin signaling. These effects were largely reversed with IL-1β antagonism (Gao et al., 2014).
An important cytokine induced by IL-1β is IL-6, which is, thus, widely considered pro-inflammatory. However, some evidence indicate that IL-6 is necessary for metabolic health. Some of those studies show that IL-6 deficiency led to development of obesity, liver inflammation and insulin resistance even during low-fat feeding (Wallenius et al., 2002; Matthews et al., 2010). Experimental evidence show that IL-6 produced by adipocytes is deleterious, while myeloid and muscle cells production of IL-6 is protective in obesity, possibly explaining the discrepancy between its inflammatory nature and metabolic importance (Han et al., 2020).
Early reports showed that macrophage TNFα contributes to insulin resistance and its deletion alleviates insulin resistance (Stephens and Pekala, 1991; Hotamisligil et al., 1993; De Taeye et al., 2007). However, it was recently reported that the deletion of macrophage TNFα did not influence insulin resistance and hepatic lipid accumulation in obese mice, indicating that macrophages are not the source of TNFα that causes metabolic dysfunction (Aladhami et al., 2021).
Although macrophages have been found to produce pro-inflammatory cytokines that enhance insulin resistance, experimental evidence demonstrate that macrophages also secrete and respond to anti-inflammatory cytokines, enhancing insulin sensitivity. For example, IL-13 produced by obese adipocytes was shown to inhibit inflammatory activation of ATMs and improve glucose and insulin tolerance (Kwon et al., 2014). In addition, IL-33 treatment of adipocytes and ATMs induces the production of IL-5, IL-6 and IL-13 which can exert protection against insulin resistance. Furthermore, IL-33 administration reduces adiposity, while also attenuating insulin and glucose intolerance caused by obesity (Miller et al., 2010). IL-10 has also been found to suppress the production of pro-inflammatory cytokines and protect from TNFα-induced insulin resistance in obesity (Lumeng et al., 2007). Moreover, IL-10 overexpression increases insulin sensitivity, protects skeletal muscle from obesity-associated macrophage infiltration and decreases production of inflammatory cytokines (Hong et al., 2009).
Lipid mediatorsPlasma and adipose tissue concentration of SPMs, leukotrienes and prostaglandin are markedly changed in human obesity and diabetes, compared to lean healthy conditions (Claria et al., 2013; Titos et al., 2016; Schulte et al., 2020). Chronic inflammation in obesity is a consequence of the failure to actively resolve inflammation, possibly due to reduction in lipid mediators. For instance, HFD increases palmitic acid, the major free fatty acid released by adipocytes, causing upregulated expression of Ptgs2 in macrophages. Ptgs2 knockout in myeloid cells of DIO mice leads to accumulation of fat in ATMs, an increase in monocyte recruitment and local proliferation of ATMs, as well as insulin resistance, inflammation and AT fibrosis. These data were recapitulated when myeloid cells did not express the PGE2 receptor EP4 (Pan et al., 2022). Conversely, Hellmann et al. (2013), found that in db/db and DIO mice, palmitic acid stimulates COX-2 and increases prostaglandin production, leading to accumulation of neutrophils and impair macrophage phagocytosis in a model of peritonitis. COX-2 or EP2 (prostaglandin 2 receptor) inhibition decreased neutrophil and apoptotic cell accumulation in the peritoneum of db/db mice, suggesting that COX-2/PGE2 impair inflammation resolution in obese mice. Taking these two studies into account, it seems that PGE2 is important in macrophages to promote inflammation resolution in obesity. However, PGE2 might be dominantly deleterious when activating other, yet unknown, cells.
The role of BLT-1 (leukotriene B4 receptor) in promoting trafficking of monocyte to adipose tissue and chronic inflammation in obesity has also been elucidated. HFD increases the circulating levels of monocytes that express Blt1, while its knockdown prevents the accumulation of ATMs and expression of Il6 and Ccl2. Blt1 deficiency was also associated with decreased hepatic triglycerides, leading to improved systemic glucose and insulin tolerance. This indicates that BLT-1 may contribute to obesity-induced metabolic derangement (Spite et al., 2011).
Resolvins were also shown to improve insulin and glucose tolerance. In obese mice, supplementation with either RvD1 (Hellmann et al., 2011) or RvE1 (Pal et al., 2020) improved fasting glucose and insulin levels. RvD1 treatment was further shown to improve AT inflammation and promote reparative macrophage polarization (Hellmann et al., 2011), suggesting that these resolvins might be considered as treatment for obesity-induced insulin resistance.
MicroRNAsATMs from obese animals secrete exosomes containing miRNAs that are taken up by insulin target cells, both in vitro and in vivo, which leads to cellular and systemic insulin resistance and glucose intolerance. Among these is miR-155, which inhibits insulin signaling and glucose tolerance through suppression of Pparg (Tryggestad et al., 2019). In contrast, treatment of obese mice with ATM-exosomes derived from lean mice leads to improvement in glucose tolerance and insulin sensitivity, indicating that ATM-exosomes perform both detrimental and beneficial actions depending on the metabolic state of the AT (Ying et al., 2017; Gao et al., 2021). Another miR that is packaged in exosomes and can regulate metabolic health is miR-690. It has been shown that IL-4/13 treated bone marrow-derived macrophages produce miR-690 rich exosomes. Administration of these exosomes to obese mice improves their glucose and insulin intolerance. A similar protective phenotype was observed after transferring exosomes that exclusively carry miR-690, and no other miR, to obese mice (Ying et al., 2021).
In addition to miR-690 there are other miRs that promote metabolic health. PPARγ, which is important for macrophage anti-inflammatory polarization, is responsible for the expression of miR-223. Deficiency of miR-223 in HFD-fed mice causes marked glucose intolerance and insulin resistance (Zhuang et al., 2012; Ying et al., 2015). In addition, miR-223 reduces Pknox1 levels, a protein that induces pro-inflammatory macrophage polarization (Zhuang et al., 2012). miR-17–92 family are also crucial regulators of the balance between pro-inflammatory and anti-inflammatory cytokines. HFD fed miR-17–92−/− mice had increased body weight, adiposity and TNFα but reduced IL-10 when compared with wild type mice. Treatment of miR-17–92−/− mice with anti-TNFα suppresses obesity and increases IL-10 via upregulation of Fos Proto-Oncogene expression, suggesting that miR-17–92 controls obesity by upregulating Il10 and suppressing Tnfa (Zhang et al., 2020).
In addition, HFD downregulates miR-30 expression and subsequently upregulates pro-inflammatory responses in ATMs. Enhancement of miR-30 in RAW264.7 (macrophage cell line) reduced macrophage inflammatory polarization and decreased TNFα and CCL2 production by inhibiting the Notch ligand DLL4. This suggests that enhancement of miR-30 may produce an anti-inflammatory effect and can be considered for resolution of inflammation in obesity (Miranda et al., 2018).
Efferocytosis and phagocytosisEfferocytosis is essential for successful resolution of inflammation and maintenance of tissue homeostasis. Obesity and its associated complications compromise the function of macrophages leading to prolonged and impaired resolution of inflammation. Several factors have been found to be responsible for impaired efferocytosis and phagocytosis in obesity and diabetes. c-Mer tyrosine kinase (MerTK) has been shown to play a crucial role in the recognition and subsequent ingestion of apoptotic cells by macrophages. Human diabetic hearts were shown to have reduced levels of MerTK, coupled with a decrease in miR-126. Mechanistically, hyperglycemia reduces macrophage miR-126, which cannot inhibit Adam9 expression. ADAM9 cleaves and inactivates MerTK, thereby inhibiting efferocytosis (Suresh Babu et al., 2016). Another cause of impaired efferocytosis and phagocytosis in obesity and diabetes is the altered ratio between free fatty acids and pro-resolving lipids. Ob/ob mice were shown to have more palmitic and stearic acid, and less EPA and DHA (Li et al., 2009). Interestingly, treatment of naïve macrophages with saturated fatty acids greatly impairs their efferocytic capability. In vivo, ob/ob atherogenic mice had more apoptotic cells in their plaques compared to atherogenic mice that are not on the ob/ob background, which was reversed by feeding an omega-3 rich diet (Li et al., 2009).
In addition to the effects of prostaglandins on impairment of efferocytosis in obesity [discussed above (Hellmann et al., 2013)], it was shown that obesity reduces macrophage erythropoietin (EPO), which elevates PPARγ levels, required for the macrophage pro-reparative phenotype. In a peritonitis model, treatment of obese mice with recombinant human EPO abrogates the defects in efferocytosis caused by obesity and stimulates inflammation resolution (Luo et al., 2019).
Weight loss in obesity might also improve phagocytic capabilities of ATMs. Single-Cell RNA sequencing of lean, obese and calorically restricted visceral AT immune cells has revealed a new distinct subpopulation of ATMs that is unique to weight loss. These ATMs express many phagocytosis/efferocytosis-related genes, such as Fcgr4, Axl, Mertk and Fcer1g. It was further demonstrated that caloric restriction-induced weight loss promotes the appearance of multinucleated ATMs, further supporting their increased phagocytic capacity. However, the function of these newly found ATMs remains to be understood (Weinstock et al., 2019). Although this study suggests that weight loss promotes beneficial changes to ATMs, two recent papers demonstrate that weight loss induced by switching from high-to low-fat diet does not reverse the obesity-induced inflammatory phenotype of ATMs (Caslin et al., 2022; Hata et al., 2023). Both these studies show that formerly obese ATMs produce more inflammatory cytokines than lean ones, and in comparable amounts to obese ATMs upon stimulation with lipopolysaccharide (LPS). These data suggest that obesity has long-lasting effects on the inflammatory state of macrophages, which can further exacerbate obesity comorbidities.
Another recent report elegantly characterized the immune compartment in obese mice following diet switch-induced weight loss and regain. Cottam et al. made several interesting observations. First, they did not find similar weight loss specific, Fcgr4 expressing ATMs, which might be due to several differences in the design of both studies. Some differences include changes in diet composition during weight loss (from 60% high fat diet to 10% low fat diet) and the duration of weight loss (9 weeks), while Weinstock et al. maintain the same HFD throughout but reduce food amounts by 30% for 2 weeks to allow weight loss. Second, they observed an ATM subpopulation that cycles together with weight, accumulating with obesity, disappearing in weight loss and reappearing in greater amounts with weight regain. Some of their marker genes include Stmn1, Pclaf, Saa3 and Slpi, suggesting they are proliferative and might be involved in efferocytosis, however, not much is known about these cells otherwise. It remains to be seen if these cycling cells contribute to the enhanced inflammation occurring with weight regain. Third, the data indicate that many transcriptional changes induced by obesity in resident ATMs and LAMs are not recovered upon weight loss, possibly priming them to respond more robustly upon weight regain (Cottam et al., 2022). How weight loss and regain influence the immune microenvironment in the AT is an important question that will require further investigations.
AtherosclerosisCardiovascular diseases (CVDs) are the leading cause of health concern globally, accounting for about one-third of all deaths. Atherosclerosis, one of the most dangerous CVDs, is characterized by the formation of plaques in the arteries, which ultimately leads to various forms of disease manifestation, depending on the plaque location, including coronary artery, peripheral artery and cerebrovascular disease, frequently resulting in what is commonly referred to as heart attack and stroke. The disease is initiated by the accumulation of modified-cholesterol moieties under the endothelial layer (Witztum and Steinberg, 1991), which induces the onset of a vicious inflammatory cycle. Stressed endothelial cells in the arteries respond by increasing their cell surface levels of various leukocyte adhesion molecules, such as Intercellular adhesion molecule (ICAM) - 1 (Kevil et al., 2001) and Vascular Cell Adhesion Molecule 1 (VCAM-1) (Hellmann et al., 2011), while they and other cells secrete chemokines that direct monocytes to the site of inflammation. Monocytes then adhere to endothelial ICAM-1 and VCAM-1 and transmigrate to penetrate the arterial wall. Once resident in the artery, monocytes become macrophages, working to engulf trapped LDL molecules through the upregulation of their scavenger receptors (Libby, 2002).
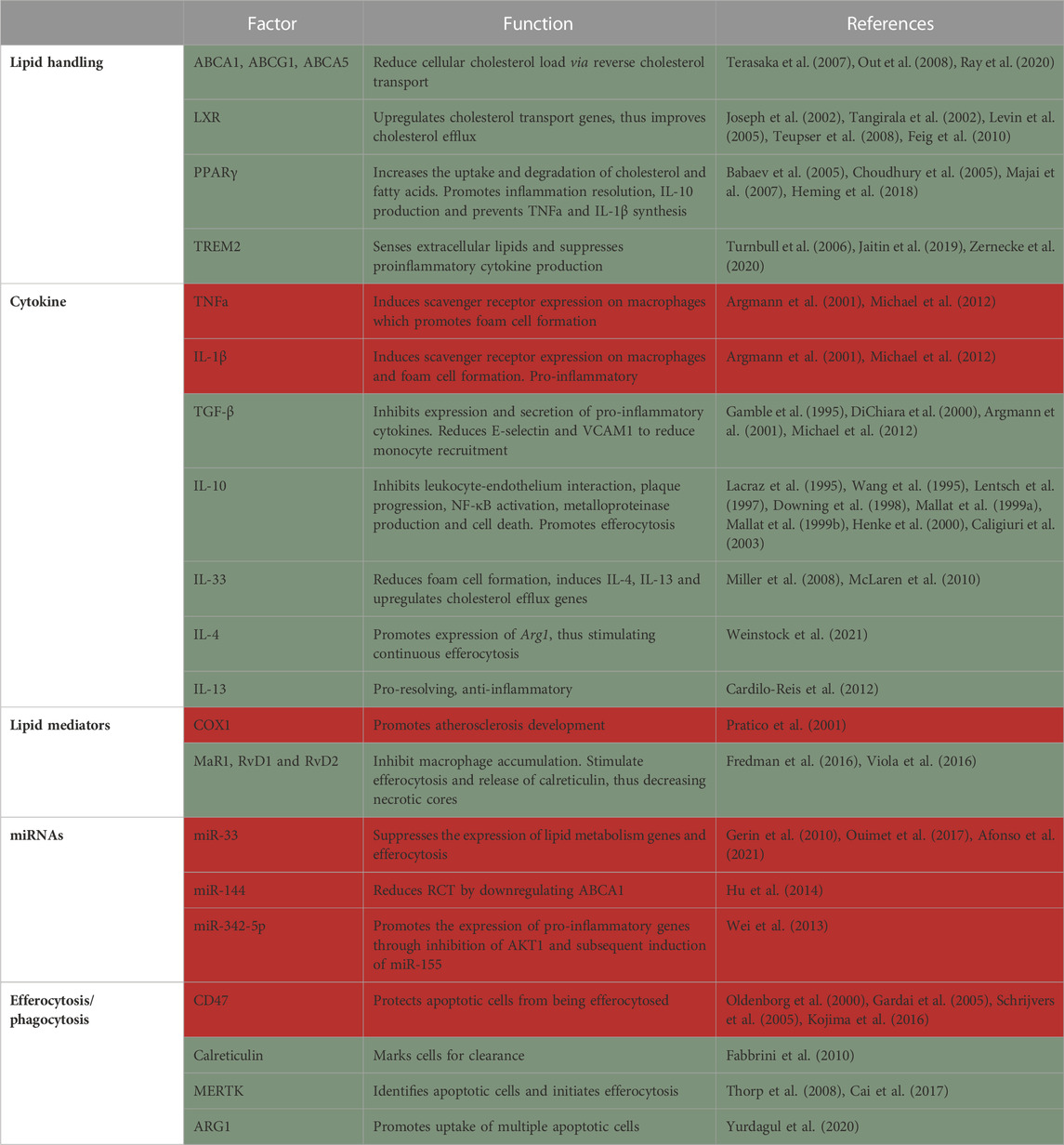
Due to the inflammatory environment, cells may undergo apoptosis. The failure to clear these dying cells by macrophages leads to secondary cellular necrosis which, over time, develops into the necrotic core, the hallmark of plaque instability. The necrotic core is also a major source of danger-associated molecular patterns, matrix metalloproteases and pro-inflammatory cytokines, which further exacerbate the disease and may ultimately lead to plaque rupture. The modern view of the role of macrophages in atherosclerosis came from pioneering studies that demonstrated macrophage presence in atherosclerotic plaques (Cookson, 1971) and the role of monocyte-derived macrophages recruitment in plaque progression (Swirski et al., 2007). With the advancements of single-cell RNA-sequencing, we have further clarity on the different plaque macrophage subtypes. Meta-analysis of plaque leukocytes described 5 major subtypes of macrophages (Cochain et al., 2018; Kim et al., 2018; Winkels et al., 2018; Lin et al., 2019; Zernecke et al., 2020). Since single-cell RNA-sequencing is continuously being collected, even more macrophage subtypes are being discovered in mice and humans (Fernandez et al., 2019; Pan et al., 2020; Eberhardt and Giannarelli, 2022), however, their distinct functions are only now beginning to be elucidated (Table 2).
TABLE 2. Summary of factors in macrophages that protect from (green) or promote (red) atherosclerosis.
Macrophage originMacrophages reside in the healthy aorta (Jongstra-Bilen et al., 2006) and were thought to be derived from both embryonic and monocytic origins (Ensan et al., 2016). However, it was recently demonstrated that unlike most tissue resident macrophages, aortic macrophages are not derived from embryonic tissues (i.e., yolk sac and fetal liver), but rather from circulating monocytes recruited to the aorta shortly after birth (Williams et al., 2020). These cells were named MacAIR and were found to proliferate locally to maintain their population. In hypercholesterolemic conditions, MacAIR form the first foam cells. MacAIR were further shown to induce the initial recruitment of monocytes to the aorta and their depletion delayed atherosclerosis initiation (Williams et al., 2020). Meta-analysis of plaque leukocytes identified lymphatic vessel endothelial hyaluronan receptor-1 (Lyve-1) as a marker for resident macrophages (Zernecke et al., 2020).
Incoming monocytes are the source of most plaque macrophages (Williams et al., 2020). Early studies implicated Ly6Chi monocytes in forming inflammatory macrophages within lesions (Swirski et al., 2007). Additionally, using knockout mouse models, several chemokines and their receptors (such as CCR2-CCL2 (Boring et al., 1998; Gu et al., 1998), CCR5-CCL5 (Veillard et al., 2004; Braunersreuther et al., 2007) and CX3CR1-CX3CL1 (Lesnik et al., 2003)] were discovered to play a major role in monocyte recruitment into the atheroma. CCR2 and CX3CR1 were shown to induce the recruitment of Ly6Chi, while CCR5 is responsible for Ly6Clo monocyte recruitment (Tacke et al., 2007). Importantly, blocking monocyte recruitment in mice via a combination of Ccl2 and Cx3cr1 double knockout with CCR5 inhibition severely impaired disease progression and reduced plaque size by 90% (Combadiere et al., 2008). Monocyte-derived macrophages were also shown to persist locally through proliferation in plaques (Robbins et al., 2013). These results raised the hope for a simple anti-atherosclerotic intervention in the form of inhibition of monocyte recruitment. However, this is more complicated than expected, due to the dichotomous role of macrophages in causing and resolving inflammation. Of importance, resolution of atherosclerosis was completely abrogated in CCR2, but not CCR5-deficient mice (Rahman et al., 2017). This suggests that while serving as instigators of atherosclerosis development and progression, monocytes and in particular the Ly6Chi subset, are indispensable for atherosclerosis resolution.
Another interesting source of cells that are perceived as macrophages in atherosclerotic plaques is vascular smooth muscle cells (SMCs). In hypercholesterolemic conditions, SMCs may show features of transdifferentiating to macrophage, downregulating SMC-specific markers and increasing their expression of macrophage markers (Andreeva et al., 1997; Rong et al., 2003; Mulvihill et al., 2004; Allahverdian et al., 2014; Shankman et al., 2015) (reviewed in (Doran et al., 2008; Orr et al., 2010; Bennett et al., 2016)]. Their function is not completely understood yet, and they will not be expanded on further in this review.
Lipid handlingAnother critical role of macrophages in the atheroma is to assist in the clearance of excess cholesterol and deliver it to the liver for redistribution, breakdown or excretion. This process is termed reverse cholesterol transport (RCT). Apolipoprotein A1, the signature protein component of HDL, interacts with cholesterol transporters such as ABCA1 (Out et al., 2008), ABCG1 (Terasaka et al., 2007) and ABCA5 (Ray et al., 2020) in various cell types (including macrophages) to facilitate the withdrawal of excess cellular cholesterol. Therefore, HDL acts as both the receiver and transporter of excess cholesterol from peripheral tissues into the liver. Additionally, HDL possesses antioxidants and anti-inflammatory potential that can aid plaque regression [reviewed in (Tall et al., 2008)]. Multiple studies have tried increasing plasma HDL levels to reduce atherosclerosis with mixed results in preclinical models and disappointing results in clinical trials. This might be because enhancement of RCT requires a coordinated response between cholesterol efflux, the capability of HDL to accept cholesterol and finally the ability of liver cells to accept it for downstream processing. Further understanding of HDL and its local functions is needed to maximize its therapeutic benefits.
RCT is induced by liver X receptors (LXRs), which upregulate the expression of various cholesterol transport genes in response to a lipid-rich environment. Transplantation of bone marrow lacking LXRα and β into ApoE−/− and Ldlr−/− mice results in aberrant regulation of cholesterol transporter genes, lipid accumulation in macrophages and increased atherosclerosis, phenotypically mimicking Tangier disease in humans (Tangirala et al., 2002). Similarly, macrophage overexpression of LXRα in Ldlr−/− mice results in reduction in lesion area at the brachiocephalic artery, while cholesterol efflux is significantly elevated (Teupser et al., 2008). Administration of non-steroidal LXR agonist (GW3965) display potent anti-atherogenic activity by inducing the expression of Abca1 and Abcg1 in Ldlr−/− mice (Joseph et al., 2002). Moreover, administration of the LXR agonist T0901317 to mice harboring existing plaques resulted in plaque regression and remodeling towards a stable phenotype. T0901317 failed to exert its anti-atherogenic effects in the absence of macrophage LXR expression (Levin et al., 2005). Furthermore, LXR is required for atherosclerosis regression since it induces CCR7-mediated egress of macrophages and dendritic cells from plaques (Feig et al., 2010). LXR further inhibits inflammatory responses by inducing macrophage phagocytic function and inhibiting macrophage proliferation (144). The loss of myeloid LXR reduces the expression of Trem2 and its related genes, thus promoting atherosclerosis progression (Endo-Umeda et al., 2022). Altogether, these studies indicate that LXR activation in macrophages has a protective effect. However, LXR can lead to hepatic fat deposition and inflammation, causing a drawback to its potential clinical use [reviewed in (Fessler, 2018)].
Another key regulator of macrophage lipid metabolism and inflammation is PPARγ. Activation of PPARγ in macrophages leads to the expression of genes involved in the uptake and degradation of cholesterol and fatty acids, as well as the synthesis of anti-inflammatory molecules such as IL-10 upon stimulation with LPS (Majai et al., 2007; Heming et al., 2018). This stimulates resolution of inflammation and prevents foam cell formation, which is a key step in the development of atherosclerotic plaques (Choudhury et al., 2005). In contrast, inhibition of PPARγ in macrophages leads to the expression of pro-inflammatory genes and the synthesis of pro-inflammatory molecules such as TNF-α and IL-1β. This promotes inflammation and foam cell formation, which contributes to the development of atherosclerotic plaques. Macrophage specific knockout of Pparg results in significantly larger plaques with more macrophages in comparison to Pparg sufficient controls. This was accompanied by increased macrophage recruitment and Ccr2 overexpression, while in vitro assays revealed reduced uptake of oxidized but not acetylated LDL (Babaev et al., 2005).
Another key regulator of lipids in macrophages is LPL. Elucidation of the role of LPL in atherosclerosis was challenging at first because homozygous knockout led to death of Lpl deficient mice soon after birth. This was overcome through transplantation of fetal liver (Babaev et al., 1999) or bone marrow hematopoietic progenitors (Van Eck et al., 2000), as well as conditional knockout models. Eventually, multiple studies showed that Lpl deficiency reduces lesion size by 30%–50% in comparison to control mice. Further experiments revealed that Lpl expression in macrophages promotes atherogenesis, as its deficiency specifically in macrophages inhibited plaque progression (Babaev et al., 1999; Babaev et al., 2000; Takahashi et al., 2013). However, Lpl macrophage-specific (Chang et al., 2019) or global (Josefs et al., 2021) deficiency in established plaques did not influence atherosclerosis regression.
In the AT, Lpl is one of the marker genes of LAMs, together with Trem2, a marker for foamy (lipid-laden) macrophages in multiple tissues (Jaitin et al., 2019; Zernecke et al., 2020). TREM2 was shown to activate the lipid associated macrophages program, acting as a sensor of extracellular lipids. Early studies demonstrated that TREM2 is expressed on infiltrating macrophages but not on myeloid progenitors, circulating monocytes or tissue-resident macrophages and TREM2 suppresses proinflammatory cytokine production (Turnbull et al., 2006). Trem2−/− macrophages were recently shown to be impaired in their lipid uptake and, thus, ability to become foam cells. Trem2 macrophage deficiency promotes their death and reduces proliferative capacity in plaques. Mechanistically, Trem2−/− macrophages have dysregulated cholesterol sensing and efflux mechanism making them susceptible to lipotoxicity-mediated cell death (Patterson et al., 2022). A study comparing serum TREM2 levels in coronary artery disease patients with healthy controls suggests that higher soluble TREM2 levels can be used as a biomarker for plaque rupture and predictor of cardiovascular death (Cuciuc et al., 2022).
CytokinesSeveral inflammatory cytokines blocking agents are currently on trial as therapy for atherosclerosis (Ridker et al., 2018; Nidorf et al., 2019; Ridker et al., 2019). The CANTOS clinical trial, featuring a monoclonal antibody against IL-1β to reduce inflammation in high-risk atherosclerosis patients did show a ∼15% decrease in recurring cardiovascular events (Ridker et al., 2017). Generally, pro-inflammatory cytokines increase cholesterol uptake, reduce efflux, promote apoptosis and impair efferocytosis. On the opposite end of the spectrum, anti-inflammatory cytokines act by reducing the cholesterol uptake and/or increasing its efflux, promoting the expression of pro-resolving proteins and mediators and inducing efferocytosis.
Anti-inflammatory cytokines can reduce cholesterol uptake in several ways. First, they inhibit pro-inflammatory cytokines, such as TNFα and IL-1β. These pro-inflammatory agents can stimulate the expression of scavenger receptors on macrophages (Hashizume and Mihara, 2012), facilitating the uptake of cholesterol and promoting foam cell formation. Anti-inflammatory cytokines, such as transforming growth factor (TGF) β, can inhibit the expression and secretion of pro-inflammatory cytokines, leading to a reduction in the expression of scavenger receptors and the uptake of cholesterol (Argmann et al., 2001; Michael et al., 2012). Second, anti-inflammatory cytokines prevent the recruitment of inflammatory cells to the arterial wall. For example, TGF-β has been shown to reduce the expression of E-selectin in endothelial cells (DiChiara et al., 2000) and VCAM1 in SMCs (Gamble et al., 1995), which are known to help immune cells bind to the endothelium and penetrate the sub-endothelial space. IL-10 also exerts its effects through inhibiting leukocyte-endothelium interaction, thus hindering their recruitment to the tissue (Downing et al., 1998; Henke et al., 2000).
Additionally, IL-10 is expressed in both early and advanced human plaques and inhibits plaque progression and rupture, NF-κB activation, metalloproteinase production and cell death (Lacraz et al., 1995; Wang et al., 1995; Lentsch et al., 1997; Mallat et al., 1999a; Mallat et al., 1999b). IL-10 acts to dampen the pro-inflammatory mediators at both transcriptional and posttranslational levels. Transgenic mice over-expressing IL-10 show reduced atherosclerotic plaques, while IL-10 deficient mice have significantly worse atherosclerosis compared to WT controls (Caligiuri et al., 2003). IL-10 was further shown to induce rearrangement of the cytoskeleton and promote apoptotic cell engulfment/efferocytosis (Proto et al., 2018). A very recent study revealed the role of macrophage derived IL-10 in limiting atherosclerosis. The authors used a combination of single-cell RNA-sequencing and flow cytometry to identify inflammatory and resident plaque macrophages as the primary source of IL-10. Myeloid specific deletion of IL-10 results in significantly larger atherosclerotic plaques along with elevated serum CCL2 and TNFα (Orecchioni et al., 2022).
IL-33 is another cytokine that seems to have a protective effect in atherosclerosis. Its levels are increased in plaques, but it is also present in normal aorta (Miller et al., 2008). IL-33 treatment was shown to reduce foam cell formation in vivo, while in vitro it reduced acetylated and oxidized LDL uptake by downregulation of Cd36 and enhanced cholesterol efflux by upregulation of Apoe, Abca1 and Abcg1 among others. IL-33 promotes the expression of other anti-inflammatory cytokines, such as IL-4 and IL-13 (McLaren et al., 2010).
The Th2 cytokines IL-4 and IL-13 promote an anti-inflammatory macrophage phenotype through the transcription factor STAT6, and were, thus, proposed as therapies for atherosclerosis. However, similarly to other cytokines, their role is complex in disease progression and resolution. Indeed, some studies reported halted plaque progression in Il4 single (King et al., 2002) and Il4/Il13 double knockouts (Weinstock et al., 2021), compared to sufficient mice. In contrast, Binder and colleagues (Cardilo-Reis et al., 2012) demonstrated that Il13-deficiency promotes atherosclerosis and that pharmacological treatment with IL-13 induces pro-resolving macrophage polarization in plaques. These results suggest that IL-4 contributes to plaque growth, while IL-13 inhibits atheroprogression. While IL-4 seems detrimental for atherosclerosis progression, it was recently shown to be required for disease resolution (Weinstock et al., 2021). In this study we demonstrated that IL-4, but not IL-13, accumulates in plaques during disease progression, but in levels that are not sufficient to promote macrophage pro-resolving polarization. With lipid lowering, there was upregulation of the Wnt signaling pathway and PGE2 production, which prime macrophages to low, plaque relevant levels of IL-4. Together, Wnt and IL-4 induce macrophage pro-resolving polarization and atherosclerosis resolution (Weinstock et al., 2021).
Lipid mediatorsLow levels of SPMs were reported in plasma of patients with chronic inflammatory diseases compared to healthy controls [reviewed in (Fredman and Tabas, 2017)]. Several SPMs have been directly implicated in improving atherosclerotic plaque inflammation (Gerlach et al., 2020). Along these lines, increased SPMs in human monocyte-derived macrophages by omega-3 supplements were linked with an upregulation of macrophage phagocytosis and a decreased uptake of oxidized LDL (Sobrino et al., 2020).
COX-1, through its products, such as thromboxane A2, has been found to promote atherogenesis and deletion of Ptgs1 (the gene encoding for COX-1) in atherogenic (Apoe−/−) mice attenuates lesion development (Pratico et al., 2001). While a dual inhibition of COX-1 and COX-2 in Ldlr−/− mice reduces lesion development as well, there is still uncertainty of the involvement of COX-2 in atherosclerosis (Pratico et al., 2001). Some studies show that pharmacological inhibition and hematopoietic genetic deletion of COX-2 either inhibit (Burleigh et al., 2002), does not affect (Olesen et al., 2002; Bea et al., 2003) or promotes (Rott et al., 2003) early atherogenesis (Burleigh et al., 2002). The discrepancy might be due to the inhibitor’s mechanism of action, specificity, and concentration, as well as the target cells. Direct examination with cell type-specific knockouts will help elucidate this issue.
Major products of COX-2 are prostaglandins. PGE2 acts through binding to its receptors (EP1-EP4) and can direct inflammation to either strengthen or resolve it. Its functions are dependent on its concentration, target cells and additional cues available in the microenvironment (Takayama et al., 2002; Takayama et al., 2006). Lack of EP4, but not EP2 in Ldlr−/− mice fed a Western Diet for 8 weeks results in reduction in plaque area and increases apoptosis in lesions (Babaev et al., 2008), indicating processes that both harm and protect from the damages of atherosclerosis. Contrarily another study found that EP4 deficiency does not influence early atherosclerosis. At more advanced stages (after 10 weeks of Western Diet), although EP4 deficiency failed to change lesion size, it did induce pro-inflammatory gene expression (Tang et al., 2011). On the other hand, others have proposed that PGE2 may cooperate with IL-4 in macrophages to stimulate resolution of atherosclerosis (Weinstock et al., 2021).
SPMs were also shown to improve atherosclerosis. MaR1 and RvD2 were found to inhibit the progression of atherosclerosis by halting the increase in macrophage and necrotic core content in Apoe−/− mice (Viola et al., 2016). Others have found a marked difference in SPMs levels in vulnerable plaques compared with stable ones, including
留言 (0)