
記住我
Cancer, an intricate disease characterized by uncontrolled cell proliferation and evasion of regulated cell death mechanisms, is a significant global health concern (Brown et al., 2023; Bhat et al., 2024). Among the several cellular mechanisms disrupted in cancer, the regulation of cell death pathways is crucial (Peng et al., 2022; Tong et al., 2022; Gong et al., 2023; Hadian and Stockwell, 2023). Programmed cell death (PCD), also known as regulated cell death (RCD), is a genetically controlled process in which cells die in an orderly manner (Koren and Fuchs, 2021; Gong et al., 2023). RCD encompasses several mechanisms, including apoptosis, necroptosis, autophagy and the newly identified pathways of pyroptosis, ferroptosis, cuproptosis, and parthanatos (Galluzzi et al., 2018) (Figure 1). Each of these mechanisms is crucial for maintaining cellular balance and responding to cellular stress (Tang et al., 2019; Lamichhane and Samir, 2023). When mammalian cells experience irreversible disruptions in their internal or external milieu, they can initiate several signal transduction cascades that ultimately result in cell death (Kayagaki et al., 2024; Newton et al., 2024). In cancer, the disruption of these pathways not only enables the initiation and progression of tumors but also significantly affects treatment resistance and patient outcomes (Table 1) (Gong et al., 2023). Each of these RCD patterns is triggered and propagated through molecular pathways that exhibit significant connectivity (Tang et al., 2019) (Figure 2). Each variant of RCD exhibits a diverse array of morphological characteristics, ranging from complete to partial programmed cell death, which elicit unique immunomodulatory properties, including anti-inflammatory effects, promotion of immune tolerance, enhancement of inflammation, and immunogenicity. Apoptosis, marked by regulated cell shrinkage and membrane blebbing, typically leads to anti-inflammatory outcomes since apoptotic cells are phagocytosed without provoking immune activation (Elmore, 2007). Autophagy is a process of cellular degradation that generally promotes cell survival; however, under prolonged stress, it can result in cell death. Autophagy can either suppress or promote inflammation based on the context, as it regulates the immune response by degrading immune modulators or releasing signals that activate immune cells (Liu et al., 2023). Necroptosis, characterized by membrane rupture and the release of cellular contents, triggers inflammation by activating immune cells via damage-associated molecular patterns (DAMPs) (Kaczmarek et al., 2013). In a similar manner, pyroptosis, characterized by pore formation and cell lysis, enhances inflammation through the release of pro-inflammatory cytokines such as IL-1β (Liu Y. et al., 2024). Cuproptosis, a form of cell death that relies on copper, inflicts damage on the mitochondria and has the potential to trigger immune responses, although its specific immunomodulatory characteristics are still under investigation (Springer et al., 2024). Ferroptosis, initiated by iron-dependent lipid peroxidation, has the potential to promote inflammation via the release of DAMPs, which in turn can affect immune responses (Qi and Peng, 2023). Parthanatos, resulting from excessive PARP activation that leads to significant DNA damage, can trigger inflammation while potentially fostering immune tolerance in chronic conditions (Huang et al., 2022). In summary, these RCD pathways influence immune dynamics by either inhibiting or facilitating immune activation, thereby affecting cancer progression and treatment results. (Galluzzi et al., 2018; Liao M. et al., 2022).
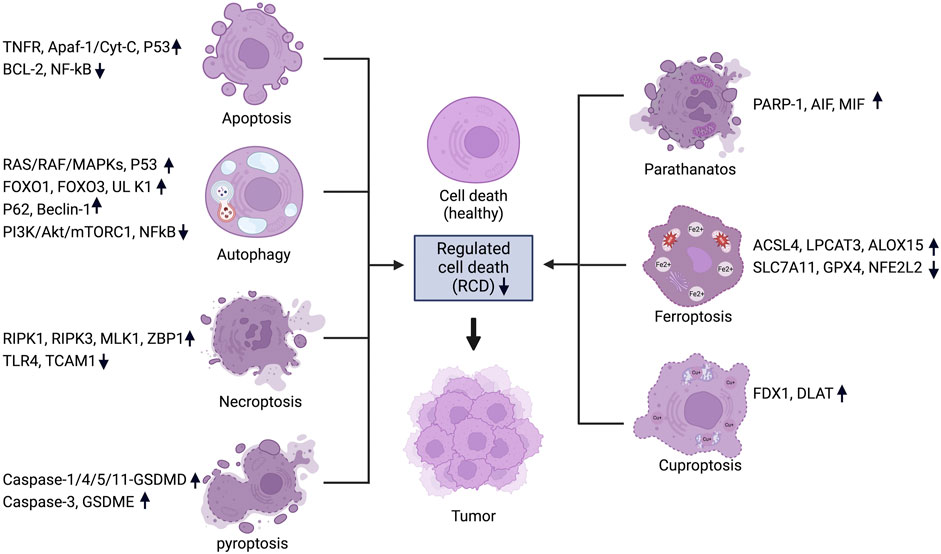
Figure 1. Regulated cell death pathways in cancer and their associated genes. For each RCD pathway, a set of key regulators are listed and the change of expression levels are indicated. Figure created using Biorender.
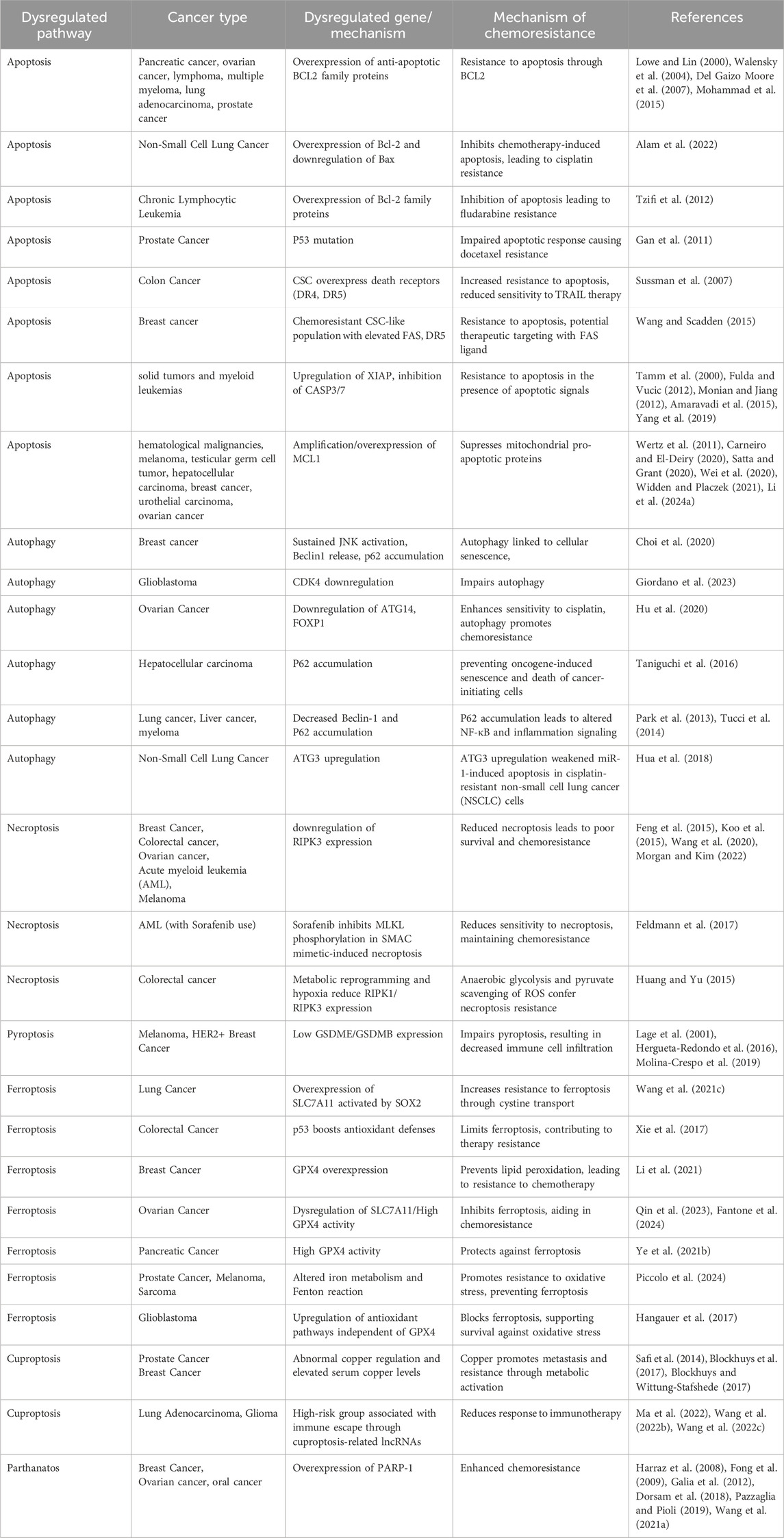
Table 1. Role of RCD pathways in cancer.
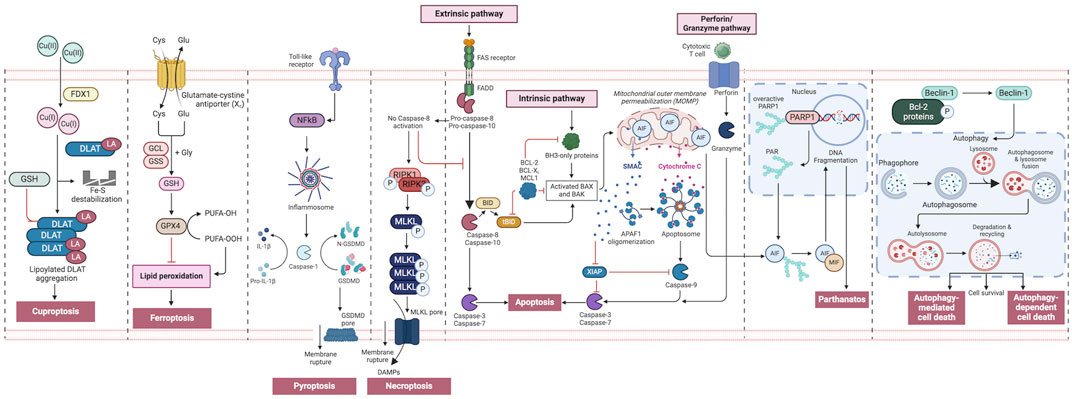
Figure 2. An overview of regulated cell death processes. A summary of the RCD pathways involved in cancer pathogenesis. Intrinsic apoptosis: Following an inherent fatal signal, BH3-only proteins activate BAX and BAK either directly or indirectly by binding to and blocking BCL-2 proteins. The mitochondrial outer membrane is then permeabilized (MOMP), releasing cytochrome C (Cyt C) and SMAC, the latter of which can suppress apoptosis. The apoptosome is subsequently produced, which activates caspase-9, followed by caspases 3 and 7, and initiates apoptosis. Extrinsic apoptosis: When death receptors (TNFR1, FAS, or TRAIL-R) receive an extrinsic fatal signal, they join with pro-caspase-8 and -10 to create complex I. Complex IIa is then generated, resulting in caspase-8 and -10 activation. Apoptosis is then initiated either directly by cleaving caspases-3 and -7, or indirectly by cleaving BID into tBID and activating BAX and BAK. Granzyme pathway: Cytotoxic T-cells are the main controllers for the granzyme pathway, which results in caspase-10 activation that in turn activates caspase-3. Granzyme B can activate caspases in the targeted cell. Necroptosis occurs when an extrinsic fatal signal is received but caspase-8 is not activated. Complex IIb (also known as the necrosome) is generated. This causes RIPK 1 and 3 to phosphorylate and activate mixed lineage kinase domain-like pseudokinase (MLKL). MLKL then forms a complex, causing the release of cytokines, chemokines, and damage-associated molecular patterns (DAMPS). Finally, this causes inflammation and necroptosis of the cell. Pyroptosis occurs when toll-like receptors (e.g., TLR4) detect an external fatal signal. Nuclear factor kappa B (NF-KB) signaling is initiated. This causes inflammasome development and subsequent caspase-1 activation. Then, pro-IL-1b is converted to active IL-1b, and gasdermin D (GSDMD) is broken down into N-GSDMD fragments resulting in inflammation and pyroptosis of the cell. Parthanatos occurs when an inherent fatal signal arises (for example, high reactive oxygen species accumulation), poly [ADP-ribose] polymerase 1 (PARP-1) is activated. Overactivation of PARP-1 can result in the accumulation of PAR polymer and the translocation of apoptosis inhibitory factor (AIF) from the mitochondria. AIF forms a compound with macrophage migration inhibitory factor (MIF) and re-enters the nucleus. Ultimately, this leads to cell parthanatos and DNA fragmentation. Autophagic Cell Death: Beclin-1 generally forms a complex with Bcl-2 proteins. After they have been phosphorylated and inactivated, free Beclin-1 can start autophagy. Ferroptosis occurs exclusively when there is an imbalance in the regulatory system, leading to the accumulation of lipid peroxide to a lethal threshold. Transferrin (TF) binds to extracellular Fe3+ and facilitates its transport into cells via transferrin receptor 1 (TfR1), where it is then converted to Fe2+. Later, intracellular divalent metal transporter 1 (DMT1) and zinc transporter 8/14 (ZIP8/14) store the Fe2+ in the intracellular labile iron pool (LIP). Fe2+ transfers electrons through the Fenton reaction with peroxide, resulting in the production of oxidizing free radicals. Following an excessive accumulation of iron within cells, numerous free radicals interact with polyunsaturated fatty acids (PUFA) found in the phospholipids of cell membranes resulting in the formation of lipid peroxides, which ultimately lead to cell death. The intracellular antioxidant stress system depends on GPX4 to eliminate surplus lipid peroxides. The Cystine/glutamate antiport (system xc−) facilitates the movement of glutamate from within cells to the outside, while simultaneously transporting cystine from the outside into cells. Cuproptosis: FDX1 plays a crucial role as a copper ion carrier in the induction of cell death and is involved in the regulation of protein lipoylation. Elevated copper levels foster the accumulation and functional impairment of lipoylated proteins, leading to instability of iron–sulfur cluster proteins, protein toxicity stress, and ultimately cell death. In addition, excessive copper binds to lipoylated DLAT, triggering abnormal oligomerization of DLAT and the formation of DLAT foci. This process contributes to cellular protein toxicity stress, further exacerbating cell death. Figure created using Biorender.
Targeting various RCD pathways to treat cancer has been under intensive investigation for several decades (Peng et al., 2022). Research in the last decade has revealed novel RCD pathways and with these discoveries, progress has been made in clinical application to target these newly identified pathways for cancer treatment (Man et al., 2017; Seehawer et al., 2018; Zhou et al., 2021; Wang Y. et al., 2022; Zhang C. et al., 2022). Furthermore, therapeutic approaches that target these RCD pathways have been used in combination with immunotherapeutic agents to further enhance their efficacies (Tong et al., 2022). Such combined approaches have the potential to significantly improve patient outcomes. Despite notable advancements, major challenges such as treatment resistance exist. This review summarizes recent advancement in preclinical and clinical development to target RCD pathways in cancer from a therapeutic standpoint, exploring how alterations in these mechanisms contribute to cancer development and impact the efficacy of current treatment methods.
1.1 Dysregulated apoptosis in cancer and targeting strategies for therapyApoptosis is a vital intracellular mechanism that maintains tissue homeostasis in an organism by regulating cell populations (Elmore, 2007; Akhtar and Bokhari, 2024) (Figure 1). However, in cancer, cells lose their capacity to undergo apoptosis-induced death, which results in unchecked cell proliferation (Morana et al., 2022). Therefore, targeting the regulation of the apoptosis signaling pathway can be one of the crucial methods to improve cancer treatment (Pfeffer and Singh, 2018). Apoptosis is characterized by cell shrinkage, chromatin condensation, membrane blebbing, DNA breakage, and apoptotic body formation (Elmore, 2007). It involves two primary pathways: the extrinsic pathway, triggered by death receptors, and the intrinsic pathway, regulated by mitochondria (Zhang et al., 2005; Jan and Chaudhry, 2019) (Figure 2). The extrinsic pathways are controlled by transmembrane death receptors belonging to the CD95 (Apo-1 or Fas)/TRAIL/tumor-necrosis factor (TNF) receptor 1 family. When death ligands such as TNFα (tumor necrosis factor-alpha), Fas ligand (FasL), or TRAIL bind to their corresponding cell surface receptors—TNFR1, Fas, and death receptors 4 and 5 (DR4/5)—it triggers a signaling cascade. This ligand-receptor interaction leads to the recruitment and activation of caspase-8, an initiator caspase, which in turn activates downstream effector caspases (Annibaldi and Walczak, 2020). The mitochondrion is involved in the other primary route that is responsible for death signaling. It performs the function of an integrating sensor of numerous death insults by releasing cytochrome c into the cytosol, where it then activates caspase. It is believed that the mitochondrial route is the primary target of survival signaling pathways (Elmore, 2007). The Bcl-2 family controls the mitochondrial (intrinsic) pathway, which is triggered by damage of the mitochondria and the subsequent release of cytochrome c. This route is initiated by cytotoxic agents and UV radiation. Cytochrome c, Apaf-1, d-ATP/ATP, and procaspase-9 interact to form an apoptosome, which then triggers the caspase cascade (Wang and Youle, 2009). Additionally, a third pathway related to endoplasmic reticulum (ER) stress has also been described (Iurlaro and Munoz-Pinedo, 2016). Stress causes mutant proteins to accumulate in the endoplasmic reticulum, disrupting the balance between protein folding and protein requirement. This event triggers the unfolded protein response (UPR), which identifies and modulates ER stress (Schroder and Kaufman, 2005; Gardner et al., 2013). Key sensors in the UPR—ATF6 (activating transcription factor 6), IRE1α (inositol-requiring enzyme 1 alpha), and PERK (protein kinase R-like ER kinase)—are activated when misfolded protein concentrations exceed a certain threshold. If the stress is too severe or prolonged, the UPR can shift from a protective role to triggering apoptosis, in order to eliminate the affected cell and prevent damage (Spencer and Finnie, 2020). Despite having distinct mechanisms of initiation, these intrinsic, extrinsic and stress-induced pathways all lead to activation of a series of proteolytic enzymes that are members of the caspase family (Elmore, 2007; McIlwain et al., 2015) (Figure 2). The caspases, which are cascades of cysteine aspartyl proteases are produced as dormant zymogens, which are then activated by proteolytic cleavage. This is normally accomplished by the action of upstream apical caspases (McIlwain et al., 2015). Apart from these intrinsic, extrinsic and stress-induced processes, there exists an additional pathway that entails T cell mediated cytotoxicity and perforin/granzyme-dependent cell death. The cell death inducing enzymes in this pathway are granzyme B and granzyme A proteases (Trapani and Smyth, 2002).
Cancer cells often overexpress proteins that prevent the apoptotic cascade from being activated, including Bcl-2 and related anti-apoptotic proteins such as Bcl-xL, Mcl-1, A1/Bf1 and Bcl-w (Table 1) (Lowe and Lin, 2000). Targeting these proteins has become a strategy to inhibit cancer proliferation and promote cell death (Frenzel et al., 2009; Carneiro and El-Deiry, 2020). Developing cancer drugs targeting the apoptosis pathway represents the first phase of clinical development in the field (Jan and Chaudhry, 2019). A comprehensive list of compounds targeting apoptotic pathways and demonstrating anti-cancer properties is presented in Table 2. ABT-737 was the initial chemical inhibitor targeting Bcl-2, Bcl-xL and Bcl-w (Del Gaizo Moore et al., 2007). It binds to the hydrophobic pocket of Bcl-2 family members and has shown efficacy against lung cancer, especially when combined with chemotherapy and radiation therapy. Navitoclax (ABT-263) demonstrates anti-cancer properties, particularly when used with MEK or tyrosine kinase inhibitors against solid tumors (Tse et al., 2008; Walensky et al., 2004) (ABT-199), a potent Bcl-2 inhibitor, has shown promising outcomes for treating acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL) and non-Hodgkin lymphoma (NHL) (Souers et al., 2013). Selective Bcl-xL inhibitors include a vaccine for prostate cancer and ABBV-155, an antibody-drug conjugate being studied as monotherapy or for use in combination with taxanes for solid tumors (Walensky et al., 2004).
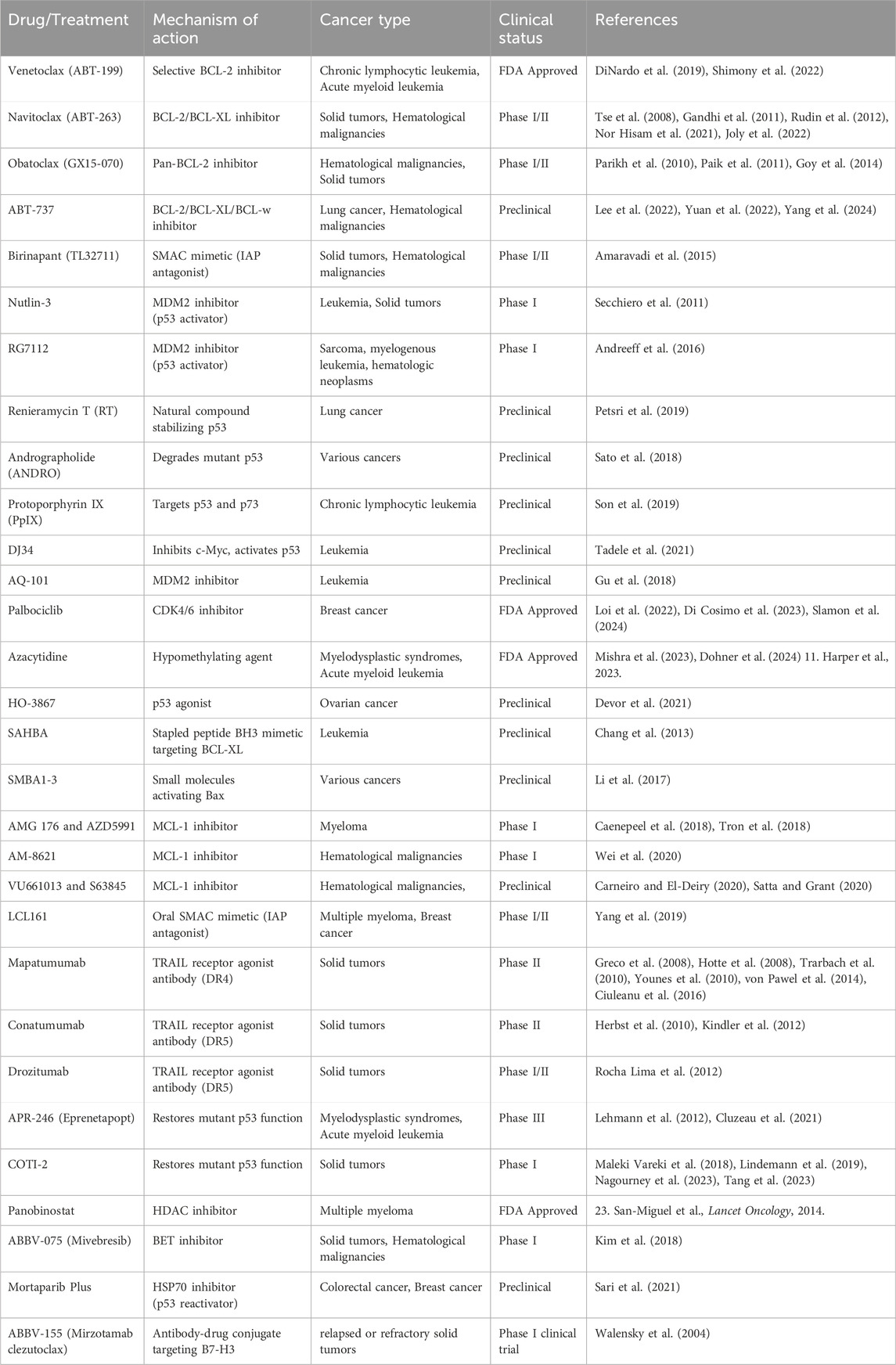
Table 2. Apoptosis targeting drugs for cancer therapy.
BH3 mimetics have been effectively created using stapled peptides that specifically bind through protein-protein interactions and have an improved ability to enter the cell (Ali et al., 2019). SAHBA (Stabilized Alpha-Helix of BCL-2 Domains) mimics the α-helical BH3 section of proapoptotic BID, efficiently enters leukemia cells, binds to Bcl-xL, and promotes apoptosis (Chang et al., 2013). Targeting Bax using small molecules like SMBA1-3, which bind directly to Bax and inhibit the phosphorylation of S184, promotes cytochrome c release and apoptosis (Li et al., 2017).
The Bcl-2 family member Mcl-1 can prevent apoptosis induced by multiple apoptotic triggers such as radiation and chemotherapy (Wertz et al., 2011; Widden and Placzek, 2021). AM-8621 attaches to the Mcl-1 binding pocket, displaces BIM and induces apoptosis in a myeloma cell line (Wei et al., 2020). Derivatives AMG 176 and AZD5991 have shown notable outcomes in combination with venetoclax and chemotherapy (Caenepeel et al., 2018; Tron et al., 2018). Mcl-1 inhibitors VU661013 and S63845 show promise in treating blood cancers and overcoming resistance to venetoclax when used in combination with other therapies (Carneiro and El-Deiry, 2020; Satta and Grant, 2020). In addition, IAP inhibitors have been used to target apoptosis in cancer (Fulda and Vucic, 2012; Monian and Jiang, 2012). Antagonists like LCL161 and birinapant (TL32711) show promising anti-tumor effects, particularly in combination with chemotherapy, radiation and the immune checkpoint inhibitor (ICI) anti-PD1 pembrolizumab (Amaravadi et al., 2015; Yang et al., 2019).
Agonist antibodies were also created targeting DR4 and DR5 due to their favorable half-life and notable preclinical efficacy (Hymowitz et al., 1999; LeBlanc and Ashkenazi, 2003). The only clinically tested anti-DR4 monoclonal antibody is mapatumumab, a completely human DR4-agonistic antibody with selective and strong binding to DR4 and high cytotoxicity (Pukac et al., 2005). Mapatumumab was tested in phase I and II clinical trials for HCC, NSCLC, colorectal cancer, and refractory non-Hodgkin’s lymphoma (Greco et al., 2008; Hotte et al., 2008; Trarbach et al., 2010; Younes et al., 2010; von Pawel et al., 2014; Ciuleanu et al., 2016), but none of the assays met the initial objectives, ending clinical development. Unlike DR4, several DR5 agonist antibodies have been developed and tested in clinic including Conatumumab, Drozitumab, Lexatumumab, LBY135, Tigatuzumab, and DS-8273a (Belyanskaya et al., 2007; Herbst et al., 2010; Kang et al., 2011; Forero-Torres et al., 2013; Burvenich et al., 2016; Dominguez et al., 2017; Forero et al., 2017). Conatumumab and Drozitumab demonstrated efficacy in advanced solid tumors, while Lexatumumab was tested in prostate and bladder cancer cells (Shimada et al., 2007; Herbst et al., 2010; Rocha Lima et al., 2012). DS-8273a is the newest clinically tested anti-DR5 antibody. The initial study showed that DS-8273a might be used to eliminate myeloid-derived suppressor cells in advanced cancer patients, but no objective response was seen (Dominguez et al., 2017). It is tested in three more clinical trials to assess its safety in advanced solid tumors and lymphomas or its efficacy in combination with Nivolumab in advanced colorectal cancer and unresectable stage II and IV melanoma (Dubuisson and Micheau, 2017). In addition, chimeric mouse–human antibodies LBY135 and Tigatuzumab were developed. Solid advanced cancers tolerated LBY135 well, and Tigatuzumab was investigated for relapsed lymphoma or solid malignancies (Forero-Torres et al., 2010; Sharma et al., 2014). Conatumumab and Drozitumumab reached phase II clinical trials, but Lexatumumab, LBY-135, and Tigatuzumab did not (Dubuisson and Micheau, 2017).
The p53 protein, a critical tumor suppressor, is often altered or deactivated in various malignancies, making it an ideal target for therapeutic treatments. Several p53-targeted medicines have been developed to help restore or improve p53 function. MDM2 inhibitors, including Nutlin-3, APG-115, RG7388, DS-3032, and MK-8242, suppress the p53-MDM2 interaction, stabilizing p53 and inducing apoptosis in malignancies such as gastric cancer and leukemia (Ding et al., 2013; Levine, 2022). Other MDM2 antagonists include AMG-232, HDM201, BI 907828, and ALRN-6924 (Carneiro and El-Deiry, 2020; Peng et al., 2022). MDMX inhibitors, such as XI-011 and DIMP53-1, restore p53 stability by inducing apoptosis and reducing migration in cervical and colon malignancies (Soares et al., 2017; Zhang J. et al., 2022). Small compounds such as PRIMA-1 (APR-017), APR-246 (Eprenetapopt), and COTI-2 restore mutant p53 to a functional state, reactivating its tumor-suppressive capabilities, with potential therapeutic uses in a variety of malignancies (Berke et al., 2022). The p53 agonist HO-3867 restores transcriptional repression in mutant p53, especially in ovarian cancer, resulting in cell death (Devor et al., 2021).
Cyclophilin A (CypA) inhibitors, such as HL001, impede MDM2-mediated p53 degradation, resulting in cell cycle arrest and death in NSCLC (Lu et al., 2017). Natural compounds such as Renieramycin T (RT) (Petsri et al., 2019) and Protopine (Son et al., 2019) stabilize p53, inducing apoptosis in lung and colon tumors, respectively, whereas Andrographolide (ANDRO) degrades mutant p53 (Sato et al., 2018). Actinomycin V and TCCP also increase p53 expression, which causes apoptosis in many cancer cells (Lin S. Q. et al., 2019; Rashmi et al., 2019). Heat-shock protein inhibitors, such as Mortaparib (Plus), reactivate p53 by disrupting its association with mortalin, causing apoptosis in colorectal and breast malignancies (Sari et al., 2021). Furtherrmore, Protoporphyrin IX (PpIX) targets both p53 and its homolog p73, which promotes apoptosis in CLL (Son et al., 2019).
Novel therapeutics include gold complexes like MC3, which upregulate p53 via the ROS formation and have shown effectiveness in colorectal cancer (Dabiri et al., 2019), as well as platinum-based compounds like bromocoumarinplatin 1 and diplatin, which activate p53 to overcome cisplatin or carboplatin resistance respectively in lung cancer (Lin X. et al., 2019; Ma et al., 2020). Other small compounds, such as DJ34, kill leukemia stem cells by inhibiting c-Myc and activating p53 (Tadele et al., 2021), whereas AQ-101 inhibits MDM2 to activate p53 and increase apoptosis in leukemia (Gu et al., 2018).
Research is exploring inhibitors of uncontrolled oncogenic effectors such as PI3K, AKT, β-catenin, Myc, CDKs, mTOR, and VEGF (Sever and Brugge, 2015). CDK4/6 inhibitors like palbociclib enhance cell death and induce cell cycle arrest in various cancers (Tao et al., 2016). Epigenetic strategies focused on inducing apoptosis in cancer cells involve histone deacetylase (HDAC) inhibitors and Bromodomain and Extra-Terminal motif (BET) inhibitors (Bolden et al., 2006; Kim et al., 2018). HDAC inhibitors, such as panobinostat, enhance Noxa expression, reduce Mcl-1 levels, and increase sensitivity to Bcl-2 inhibitors (Liu et al., 2018). They also enhance the effectiveness of MEK inhibitors and venetoclax in treating multiple myeloma (DiNardo et al., 2019). BET inhibitors like ABBV-075, when combined with venetoclax, demonstrate promising outcomes in patients with cutaneous T cell lymphoma (CTCL) (Kim et al., 2018). The hypomethylating agent azacytidine, when combined with venetoclax and ABT-737, has shown promising results (Mishra et al., 2023).
1.2 Targeting autophagy for cancer therapyAutophagy, a critical mechanism for maintaining cellular balance by removing damaged organelles and protein aggregates, can also facilitate cell death (Liu et al., 2023). Cells can undergo autophagy-related cell death in two primary ways: autophagy-dependent cell death (ADCD) which transpires independently of other programmed death mechanisms and autophagy-mediated cell death (AMCD) that occurs when autophagy-related molecules directly engage with those implicated in forms of cell death. (Zhou et al., 2022) (Figure 1). Moreover, autophagy is linked to other cell death mechanisms, such as apoptosis, necrosis, and ferroptosis, through a variety of processes (Dunkle and He, 2011; Gordy and He, 2012; Chen et al., 2018; Liu J. et al., 2020; Peng et al., 2022) (Figure 2).
Three distinct forms of autophagy have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (Parzych and Klionsky, 2014). Microautophagy is a form of autophagy in which lytic organelles autonomously engulf and degrade cytoplasmic components. It is essential for regulating biosynthesis, transport, metabolic adaptability, organelle remodeling, and the maintenance of cellular component quality (Saha et al., 2018). Macroautophagic autophagosomes convey cellular constituents for destruction to endosomes or lysosomes (Feng et al., 2014). Autophagy starts with an isolating membrane known as the phagophore, which encases a portion of the cytoplasm. The Atg9 protein promotes growth by supplying crucial lipid constituents. The Atg1 and Atg9 proteins, together with a phosphatidylinositol 3-kinase complex, govern this activity. In the subsequent phase, two conjugation steps transpire. The first activation entails Atg12 and the Atg7 protein. The Atg12 protein is conveyed to the Atg10 protein, leading to a covalent connection with Atg5. The Atg12-Atg5 complexes subsequently associate with the Atg16L protein. The ATG12-ATG5-ATG16L1 complex is essential for the production of autophagosomes. The second step of conjugation involves the proteins Atg3, Atg4, Atg7, and LC3. The Atg4 protease cleaves proLC3, resulting in the formation of LC3-I. Subsequently, the Atg7, Atg3, and Atg12-Atg5-Atg16L proteins are conjugated. The LC3-I protein interacts with the lipophilic phosphatidyle-thanolamine (PE) to generate the LC3-II form. These stages generate the autophagosome, which encapsulates a segment of the cytoplasm and proteins. The outer membrane of the autophagosome fuses with the lysosome to form an autophagolysosome. Lysosomal enzymes facilitate the digestion of the autophagolysosome’s inner membrane and its contents (Gomez-Virgilio et al., 2022; Liu et al., 2023). Chaperone-mediated autophagy (CMA) removes damaged proteins during fasting or oxidative stress. The chaperone complex links the protein’s target motif to facilitate lysosome trafficking. In the lysosome, the complex interacts with LAMP-2A’s cytoplasmic tail and is destroyed (Bejarano and Cuervo, 2010).
Autophagy plays a complex role in cancer, acting as both an inhibitor and promoter of tumor growth (Chavez-Dominguez et al., 2020; Nawrocki et al., 2020; Debnath et al., 2023). It can help cancer cells avoid damage induced by chemotherapeutics and promote chemoresistance (Table 1) (Nawrocki et al., 2020; Debnath et al., 2023). Preclinical research using chemotherapeutics like cyclophosphamide, imatinib, and vorinostat has shown that autophagy reduces the effectiveness of these drugs and contributes to acquired resistance (Mele et al., 2020). Furthermore, autophagy aids cancer cells in adapting to chemotherapy (Ahmadi-Dehlaghi et al., 2023).
Autophagy inhibitors have shown promise in combination with chemotherapeutic and targeted immunotherapeutic drugs (Table 3). While many prospective autophagy inhibitors are being developed, chloroquine (CQ) and its derivative hydroxychloroquine (HCQ) are the only approved drugs (Wang et al., 2011). HCQ, like CQ, suppresses autophagy by blocking lysosomal acidification and autophagosome degradation but has lower toxicity (Sui et al., 2013; Cook et al., 2014; Pellegrini et al., 2014; Lee et al., 2015). A Phase II trial for muscle-invasive bladder cancer is investigating the combination of HCQ with gemcitabine and cisplatin for systemic chemotherapy (Ojha et al., 2016). Similarly, in breast cancer, HCQ combined with tamoxifen was found more effective in suppressing autophagy in estrogen-positive (ER+) cell lines (Cook et al., 2014). In renal cell carcinoma, HCQ combined with temsirolimus led to increased apoptosis by inhibiting autophagy (Lee et al., 2015). Early phase I/II trials of HCQ have focused on adult solid tumors, including pancreatic adenocarcinoma, melanoma, colorectal carcinoma, myeloma, lymphoma and renal cell carcinoma, using chemotherapy drugs such as temsirolimus, bortezomib, temozolomide, vorinostat, and doxorubicin (Llovet et al., 2008; Mahalingam et al., 2014; Rangwala et al., 2014a; Rangwala et al., 2014b; Rosenfeld et al., 2014; Vogl et al., 2014; Wolpin et al., 2014). HCQ doses ranged from 400 mg to 600 mg twice daily, showing tolerability with partial responses and stable disease in some patients (Carew and Nawrocki, 2017). For advanced solid tumors and melanoma, HCQ combined with 150 mg/m2 of temozolomide showed 27% stable disease and 14% partial response in wildtype melanoma (Cheng et al., 2009; Rangwala et al., 2014b). The combination of HCQ and rapamycin, an inhibitor of mTORC1 activity, was well-tolerated in advanced solid tumor (Rangwala et al., 2014a). In myeloma, HCQ combined with bortezomib improved the efficiency of proteasome inhibitors by causing the accumulation of misfolded proteins, with 45% of patients showing stable disease. The most common adverse effects were gastrointestinal issues and cytopenias (Vogl et al., 2014).
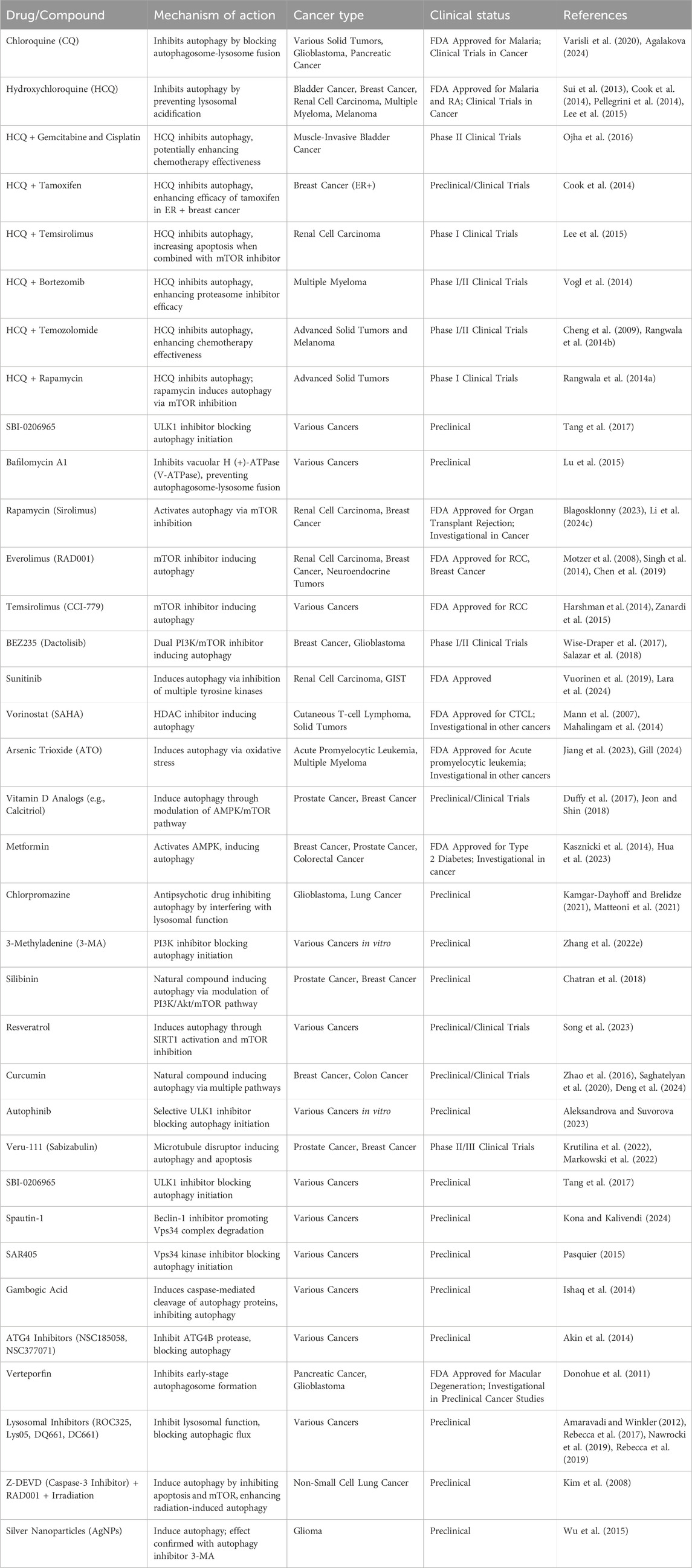
Table 3. Autophagy targeting drugs for cancer therapy.
Autophagy activation by drugs like sorafenib, a multi-tyrosine kinase mTOR inhibitor used for hepatocellular cancer, is being explored as a potential cause of drug resistance (Llovet et al., 2008). Clinical trials have investigated HCQ in hepatocellular cancer (Cheng et al., 2009) and targeted immunotherapeutic treatments, such as checkpoint inhibitors, have limited efficacy and high costs. Combining autophagy modulators like HCQ with immune checkpoint inhibitors (ICI) has the potential to improve efficacy and reduce treatment costs. Several inhibitors targeting different stages of autophagy are under investigation including inhibitors of upstream signaling molecules: SBI-0206965 (ULK1 inhibitor) (Tang et al., 2017), Spautin-1 (Beclin1 inhibitor), SAR405 (Vps18 kinase inhibitor), and gambogic acid (induces caspase-mediated cleavage of autophagy proteins) (Ishaq et al., 2014; Pasquier, 2015). Autophagy initiation inhibitors include ATG4 inhibitors NSC185058 and NSC377071, and Verteporfin (inhibits early-stage autophagosome formation) (Donohue et al., 2011; Akin et al., 2014). Lysosomal inhibitors include ROC325 (Nawrocki et al., 2019), Lys05 (Amaravadi and Winkler, 2012), DQ661 (Rebecca et al., 2017), and DC661 (Rebecca et al., 2019).
Preclinical trials have also explored autophagy’s potential to enhance radiation therapy (Kim et al., 2008; Kuwahara et al., 2011). For example, in a lung cancer mouse model, combining Z-DEVD (caspase-3 inhibitor), RAD001 (mTOR inhibitor), and irradiation induced the highest levels of autophagy and associated radiation damage. This suggests that inhibiting both apoptosis and mTOR during radiotherapy could improve outcomes in non-small cell lung cancer patients (Kim et al., 2008). Similarly, in glioma cells, autophagy induction by silver nanoparticles (AgNPs) and/or radiation was confirmed by applying 3-methyladenine (3-MA), highlighting selective autophagy as a promising therapeutic avenue for effective cancer treatment (Wu et al., 2015).
1.3 Necroptosis in cancer development and treatmentProgrammed inflammatory cell death, known as necroptosis, was first identified as an alternative to apoptosis following the activation of death domain receptors (Degterev et al., 2005; Dhuriya and Sharma, 2018) (Figure 1). Necroptosis is a regulated type of necrosis that is dependent on receptor interacting kinase-1 (RIPK1) and RIPK3 phosphorylating mixed-lineage kinase-like (MLKL) (Vandenabeele et al., 2010; Sun et al., 2012; Newton et al., 2014) (Figure 2). The necroptotic process begins when RNA- and DNA-sensing molecules and cell surface death receptors including FasRs, TNFR1, IFN receptors, and TLRs are activated (Kaiser et al., 2013). There are two ways that cell death signaling continues (Pasparakis and Vandenabeele, 2015). Complex I, a survival complex that communicates via NF-kB, can be created by TNF-α. RIPK1 deubiquitination transforms the complex into apoptotic complex IIa. When caspase-8 is absent and RIPK3 is elevated, the complex forms IIb (the necrosome). The death domain-related proteins RPK1, RPK3, and Fas on this necrosome directly phosphorylate the kinase domain-like protein (MLKL) to induce necroptosis. MLKL phosphorylation forms an oligomer that punctures the plasma membrane, killing the cell. Calmodulin-dependent protein kinase and mitochondrial serine/threonine protein phosphatase II are other RIPK3 downstream effects (He et al., 2009; Cai et al., 2014; Wang et al., 2014; Murphy, 2020). Necroptotic cell death is characterized by cell membrane perforation, elevated intracellular osmotic pressure, cell rounding and swelling, organelle swelling, impaired mitochondrial activity, mitochondrial membrane potential loss, nuclear chromatin loss, and plasma membrane rupture (Dhuriya and Sharma, 2018). Plasma membrane rupture causes potassium efflux, cytokines, and chemokines, which cause inflammation and immunological responses (Dhuriya and Sharma, 2018).
Necroptosis is involved in various aspects of tumor biology, including tumor development, necrosis, metastasis and the immune response within tumors (Najafov et al., 2017; Gong et al., 2019; Yan et al., 2022; Meier et al., 2024). This cell death pathway exhibits both pro- and anti-tumorigenic effects (Ye et al., 2023). Major regulators of necroptosis are often downregulated in cancer cells, correlating with unfavorable outcomes (Table1) (Yan et al., 2022). Necroptosis has emerged as a novel target for anticancer therapy due to its significant role in tumor biology (Gong et al., 2019).
Several natural compounds and small molecule inhibitors are known to induce necroptosis in cancer cells (listed in Table 4) (Wu et al., 2020). Chloroquine increases the expression of endogenous RIPK3 in colorectal cancer cell lines, with necroptosis being the mechanism (Meng et al., 2016). Shikonin, derived from a Chinese medicinal herb, induces necroptosis in nasopharyngeal carcinoma cells by enhancing reactive oxygen species (ROS) production and increasing RIPK1, RIPK3 and MLKL expression (Liu et al., 2019). Emodin triggers necroptosis in glioma cell lines by activating the TNF/RIPK1/RIPK3 pathway (Zhou et al., 2020). Neoalbaconol (NA), a compound derived from the fungus Albatrellus confluens, has been found to trigger necroptosis by facilitating the autocrine release of TNFα through the modulation of the RIPK/NF-κB signaling pathway and RIPK3-dependent reactive oxygen species (ROS) generation (Yu et al., 2015). The steroid glycoside Ophiopogonin D induces necroptosis in prostate cancer cells by activating RIPK1 (Lu et al., 2020). Resibufogenin inhibits colorectal cancer cell line growth by increasing RIPK3 expression (Han et al., 2018). The initiation of necroptosis can also be influenced by adjusting upstream signaling pathways, such as using the sphingosine analog FTY720 (fingolimod), which triggers necroptosis in human lung cancer cells by interacting with the I2PP2A/SET oncoprotein and activating the PP2A/RIPK1 pathway (Saddoughi et al., 2013).
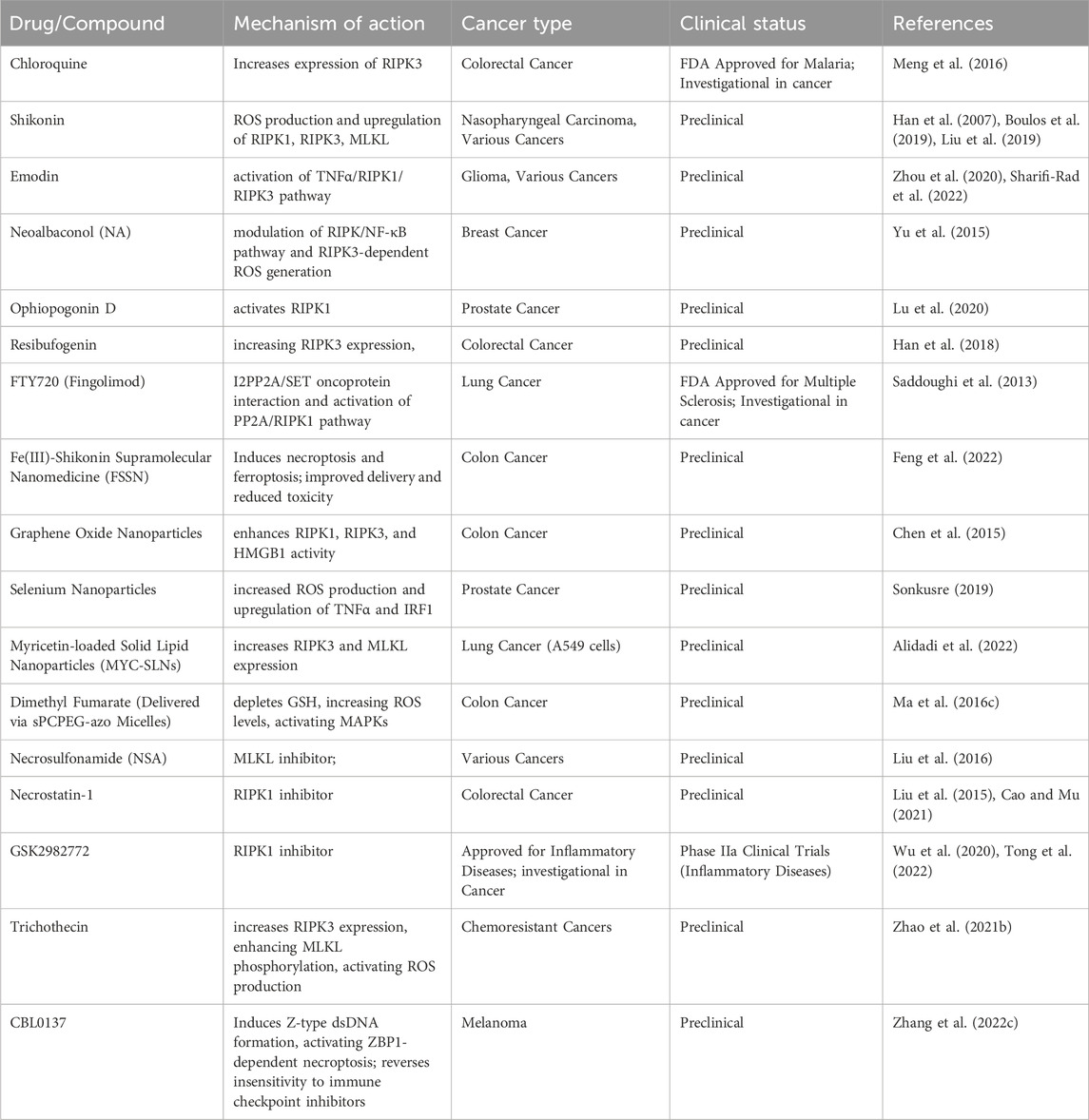
Table 4. Necroptosis targeting drugs for cancer therapy.
Nanoparticles to induce necroptosis in cancer cells is another emerging field (Mohammadinejad et al., 2019). Although the antifungal agent Shikonin shows potential, its clinical use is limited due to poor tumor specificity, low water solubility, short bloodstream half-life, and high risk of side effects on healthy tissues (Boulos et al., 2019). To address these issues, Feng et al. developed an Fe(III)-shikonin supramolecular nanomedicine (FSSN) using metal-polyphenol coordination of Fe(III) and shikonin, demonstrating improved water solubility and reduced cytotoxicity in normal cells and induced both ferroptosis and necroptosis (Feng et al., 2022). In CT26 colon cancer cells, graphene oxide nanoparticles triggered necroptosis by enhancing RIPK1, RIPK3, and HMGB1 activity (Chen et al., 2015). Similarly, selenium nanoparticles induced necroptosis in prostate adenocarcinoma cells by increasing ROS production and TNF and interferon regulatory factor 1 expression (Sonkusre, 2019). Folate-sodium alginate-cholesterol nanoparticles delivering doxorubicin and metformin achieved targeted accumulation and induced various forms of programmed cell death, including necroptosis, apoptosis, and pyroptosis in xenograft melanoma tumors (Song et al., 2021). Myricetin-loaded solid lipid nanoparticles (MYC-SLNs) enhanced necroptosis in A549 cells by increasing RIPK3 and MLKL expression without affecting apoptosis and without apparent effects on the growth and health of MRC5 cells (Alidadi et al., 2022). Ma et al. developed star-PCL-azo-PEG micelles (sPCPEG-azo) to deliver dimethyl fumarate (DMF) specifically to the colon, inducing necroptosis by eliminating GSH, increasing ROS levels and activating MAPKs (Ma Z. G. et al., 2016).
In a study conducted by Liu et al., MLKL inhibitor necrosulfonamide (NSA) was shown to significantly delay tumor growth, thus offering compelling evidence of the role necroptosis plays in promoting tumor development (Liu et al., 2016). In mice, the use of necrostatin-1, another necroptosis inhibitor, has been found to be effective in reducing colitis-associated tumorigenesis (Liu et al., 2015). There is ongoing testing of the RIPK1 inhibitor, GSK2982772, in phase 2a clinical studies for individuals with inflammatory disease. Furthermore, in a clinical trial (NCT04739618), researchers explored the potential benefits of nonablative cryosurgical freezing-induced necroptosis followed by immunotherapeutic drug injection in metastatic solid tumors. The immunotherapy included pembrolizumab (anti-PD1), ipilimumab (anti-CTLA-4), and GM-CSF. The aim was to assess the overall response rate of radiographic changes (Tong et al., 2022). In addition, induction of apoptosis has also been shown to reverse drug resistance. Xu Zhao et al., effectively employed trichothecin to trigger necroptosis in cancers that are resistant to chemotherapy. Mechanistically, the natural secondary metabolite trichothecin significantly increased the expression of RIPK3. Subsequently, RIPK3 enhanced the phosphorylation of MLKL and activated mitochondrial energy metabolism and ROS production. This novel approach sensitizes cancer cells to cisplatin therapy (Zhao X. et al., 2021).
In addition, it has been shown that necroptosis-inducing drugs could impact the effectiveness of ICIs in individuals with cancer (Tang et al., 2020). Using a viral vaccination strategy, Hoecke et al. were able to effectively deliver the necroptosis mediator MLKL to tumor cells, resulting in the promotion of necroptotic death and the enhancement of antitumor immunity. Increased immunity directly against neo-epitopes was responsible for the potent antitumor immunity (Van Hoecke et al., 2020). In addition, the RNA editing enzyme ADAR1 has been widely recognized for its role in suppressing Z-type dsRNA, a substrate for ZBP1. This suppression mechanism leads to resistance and limited responsiveness to ICIs (Zhang T. et al., 2022). However, the small-molecule drug CBL0137 has the ability to directly induce the formation of Z-type dsDNA in cells. This in turn activates ZBP1-dependent necroptosis and effectively reverses the insensitivity to ICIs in mouse melanoma models (Zhang T. et al., 2022). In addition, cIAPs have the ability to hinder the RIPK1-dependent necroptosis process. However, this inhibition can be counteracted by Smac mimetics which then trigger the activation of the necroptotic death pathway in Burkitt’s lymphoma cell lines (Koch et al., 2021). In melanoma, the response to ICIs can be enhanced by using Smac mimetics which have a direct impact on immune cells such as B cells, MDSCs, DCs, and cytotoxic T cells (Michie et al., 2020). Based on the evidence, it appears that necroptosis could potentially be employed to enhance the readiness of the tumor microenvironment for immunotherapy.
Even with progress in necroptosis research, various obstacles impede its application in cancer treatment. The practicality of necroptosis, having potential as an alternative therapy for tumors resistant to apoptosis, continues to be debated. Hitomi et al. (2008) identified a cellular signaling network that regulated necroptosis and implicated two suppressor genes, CYLD and EDD1, and four Ras-related proteins, suggesting a role in tumorigenesis. CYLD gene mutations in tumorigenic epidermal cells promote carcinoma aggressiveness by increasing angiogenic factor production, which is crucial to epidermal cancer malignancy (Alameda et al., 2010). RIPK3 and CYLD were downregulated in CLL cells, and LEF1 represses CYLD. Together, necroptosis may be crucial to carcinogenesis (Reed, 2006). Tumor heterogeneity presents a significant challenge, as numerous cancers are deficient or have mutated for essential necroptosis regulators such as RIPK3 or MLKL, which restricts the effectiveness of necroptosis inducers. RIPK3-r, a truncated splice variation of RIPK3, was dramatically elevated in colon and lung tumors compared to matched normal tissues, suggesting that it may be a primary splice form involved in carcinogenesis, according to Yang et al. (2005). The RIPK3 gene lies on chromosome 14q11.2, which is mutated in several malignancies, including nasopharyngeal carcinoma and T cell leukemia/lymphoma (Kasof et al., 2000). In non-Hodgkin lymphoma, RIPK3 gene polymorphisms increases tumor risk (Wu et al., 2012). Furthermore, existing inducers exhibit a lack of selectivity, resulting in uncontrolled inflammation and the possibility of harming healthy tissues, which raises concerns regarding off-target effects and systemic toxicity. Necroptosis may enhance anti-tumor immunity due to its pro-inflammatory characteristics, yet it also has the potential to facilitate tumor progression by creating a pro-tumor environment. Necroptosis of tumor cells can affect the TME in a way that can contribute to tumor growth because the inflammation associated with necroptosis can stimulate cell division, genetic instability, angiogenesis and metastasis (Negroni et al., 2020). Furthermore, the restricted clinical evidence, primarily based on preclinical models, along with the unpredictable nature of necroptosis outcomes, adds to the complexity of its application. Tumors can develop resistance to necroptosis, similar to how they respond to therapies that induce apoptosis. Consequently, additional research is essential to enhance targeting specificity, reduce inflammatory risks, and confirm the efficacy of necroptosis-based treatments in clinical environments.
1.4 The roles of pyroptosis in cancer cell survival and treatment strategiesPyroptosis is a form of RCD associated with inflammatory responses. It is triggered by human caspase-1, -3, -4, -5 (mouse caspase-11), −6, −8, and −9 and NLRP3 and has significant therapeutic implications for several malignancies due to its profound effects on the invasion, proliferation, and metastasis of tumor cells (Table 1) (Shi J. et al., 2014; Fang et al., 2020; Zheng and Li, 2020; Rao et al., 2022; Wei et al., 2022) (Figure 1). Gasdermin (GSDM) superfamily members, GSDMA-GSDME, which are essential to pyroptosis, are triggered by caspases and perforate the plasma membrane (Kayagaki et al., 2015; Shi et al., 2015; Rogers et al., 2019) (Figure 2). The characteristic features of pyroptosis in cancer involve gasdermins family protein cleavage and polymerization, both N-terminal and C-terminal junction domain cleavage, and activated N-terminal regions. The N-terminal generates a cell membrane pore by binding to membrane lipids, phosphatidylinositol, and cardiolipin, causing cell osmotic swelling, plasma membrane rupture, and death (Ding et al., 2016; Feng et al., 2018). Gasdermins create 10–20 nm holes in cell membranes, releasing cell contents slowly and potentially causing inflammatory responses. The cells become flattened eventually and create 1–5 μm apoptotic body-like protrusions. Nuclear concentration and chromatin DNA breaking occur as cells enlarge to rupture plasma membrane (Zhang et al., 2018). Pyroptotic pathway can occur either through classical or non-classical pathway in cancer. The classical pathway is activated by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) (Chen and Nunez, 2010; Franchi et al., 2012). Cytoplasmic pattern recognition receptors (PRRs) identify them. Based on particular inputs, nod-like receptors (NLRs) or melanoma deficiency factor 2-like receptors (ALRs) produce inflammatory bodies and activate caspase-1. After Caspase-1 cleaves GSDMD, its N-terminal aggregates into cell membrane holes (Amarante-Mendes et al., 2018; Zheng D. et al., 2020) (Figure 2). Additionally, caspase-1 cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, which are then released via the membrane hole. The nonc-lassical pyrolytic pathway requires caspase-4/-5/-11 activation. After lipopolysaccharide (LPS) stimulates the cytoplasm, caspase-4/caspase-5/caspase-11 (the human equivalent of mouse caspase-11 caspase-4/caspase-5) can directly bind to the conserved structure of LPS, lipoprotein A, causing oligomerization, activation, and the N-terminal of GSDMD to be cleaved and localized to the cell membrane to form membrane pores (Kayagaki et al., 2015). Pyroptosis is quicker and more violent than apoptosis, releasing several pro-inflammatory molecules. Cell scorch is caused by inflammatory corpuscles and GSDM family proteins.
Several chemotherapeutic drugs, including cisplatin, paclitaxel, 5-FU, lobaplatin and others have been found to trigger pyroptosis in tumor cells (Table 5) (Zhang C. C. et al., 2019; Jia et al., 2023). Chemotherapy-induced pyroptosis is frequently the result of GSDME pathway activation. Chemotherapy drug Lobaplatin triggers pyroptosis in cervical cancer and colorectal cancer by activating GSDME (Chen et al., 2022). This effect is achieved by activating caspase-3/9 through the ROS/JNK/BAX mitochondrial apoptosis pathway (Yu et al., 2019). 5-FU triggers pyroptosis in gastric cancer cells via GSDME instead of GSDMD (Wang Y. et al., 2018). When exposed to cisplatin or 5-FU, GSDME+/+ mice experience significant intestinal damage and infiltration of immune cells. On the other hand, GSDME−/− mice show less injury, indicating that triggering pyroptosis in cancer cells might offer a potential alternative approach for cancer treatment (Yu and He, 2017).

Table 5. Pyroptosis targeting drugs for cancer therapy.
Clinical trials have demonstrated that the combination of PD-L1 inhibitors with chemotherapy or radiation can effectively eliminate tumor cells through pyroptosis induction (Reck et al., 2019). This approach has shown promising results in terms of improved patient survival rates, surpassing those observed in patients solely treated with PD-L1 inhibitors. In breast cancer cells, the presence of Trimethylamine N-oxide (TMAO) can trigger GSDME-mediated pyroptosis (Wang H. et al., 2022), and when TMAO is combined with PD-1, it has the potential to enhance the antitumor effects of anti-PD-1 (Jia et al., 2023).
CAR-T cells have been successfully utilized to effectively treat hematological malignancies, yielding favorable outcomes (Gill and Brudno, 2021). However, cytokine release syndrome (CRS) is a significant side effect of this technology. When CAR-T cells release granzyme B, it can trigger pyroptosis by activating the caspase-3/GSDME pathway (Liu Y. et al., 2020). Interestingly, the elimination of GSDME through knockout has been found to effectively prevent CRS. Furthermore, the presence of perforin/granzyme B in CAR-T cells, as opposed to in CD8+ T cells, triggers GSDME-mediated pyroptosis in target cells (Liu Y. et al., 2020). These findings underscore the clinical importance of pyroptosis in immunotherapy. The release of IL-1β and IL-18 by pyroptotic cells, along with other DAMPs, can attract immune cells like dendritic cells (DCs) and macrophages (MFs) to engulf the pyroptotic cells (Wang Q. et al., 2018; Karki and Kanneganti, 2021). Mature DCs display tumor-specific antigens to activate cytotoxic T lymphocytes, which then eliminate tumors (Wang et al., 2013).
Targeted drugs have also been discovered that selectively trigger pyroptosis in tumor cells (Table 5). Val-boroPro triggers pyroptosis in primary acute myeloid leukemia (AML) cells by activating the inflammasome sensor protein CARD8 which then activates procaspase-1 (Johnson et al., 2018). In a melanoma study, it was found that the combination of BRAFi and MEKi could potentially have an antitumor effect by inducing pyroptosis through GSDME (Erkes et al., 2020). Additionally, the combination of DDP and BI2536 (a PLK1 kinase inhibitor) was observed to induce pyroptosis in esophageal cancer cells (Wu M. et al., 2019). In a study conducted by Dobrin et al., triple-negative breast cancer cells were exposed to ivermectin, resulting in the activation of the pannexin-1 pathway. This activation led to the overexpression of P2X4/P2X7 receptors, the release of ATP and ultimately the induction of pyropt
留言 (0)