
記住我
Congenital contractural arachnodactyly (CCA; OMIM 121050) was first described in 1968, and Beals and Hecht (1971) later seceded it from the Marfan syndrome (MFS; OMIM 154700), thus the naming of CCA as also the Beals–Hecht syndrome (Callewaert et al., 1993). The incidence of CCA is low, but given that its symptoms overlap with MFS, the actual percentage is hard to estimate (Tuncbilek and Alanay, 2006). CCA is a connective tissue disease. People with CCA share many distinguishing features, such as arachnodactyly, camptodactyly, crumpled ears, scoliosis, pectus deformities, flexion contractures of multiple joints (especially, fingers, elbows, and knee joints), and muscular hypoplasia (Qiu et al., 2021; Sun et al., 2022). CCA has phenotypic heterogeneity, the phenotypes of which can vary within and between families. In the most severe type, “severe CCA with cardiovascular and/or gastrointestinal anomalies”—a rare phenotype in infants with pronounced features of CCA (severe crumpling of the ears, arachnodactyly, contractures, congenital scoliosis, and/or hypotonia) and severe cardiovascular and/or gastrointestinal anomalies—it exhibits no specific geographic or ethnic predilection (Frederic et al., 2009).
CCA is caused by variants in FBN2 located on 5q23-31. FBN2 encodes fibrillin-2, a big secretory protein containing 2,912 amino acids that are expressed during early embryonic development (Peeters et al., 2021). FBN2 is related to the elasticity of the connective tissue and has been demonstrated to play an important role in the constitution of extracellular microfibrils in elastic fibers, providing strength and flexibility to the connective tissue that sustains the body’s joints and organs (Shi et al., 2013). There are other diseases that are triggered by FBN2 variants, namely, the Marfan syndrome with aortopathy, talipes equinovarus, macular degeneration, and scoliosis (Ansari et al., 2014; Buchan et al., 2014; Ratnapriya et al., 2014; Du et al., 2021).
In this study, we identified two novel FBN2 variants (NM_001999.3: c.4093T>C, p.C1365R and c.2384G>T, p.C795F) in two Chinese families diagnosed with CCA, which are likely the underlying etiologies in these two families.
Materials and methodsPatients and subjectsThis research was approved by the Review Board of Xiangya Hospital of the Central South University (approval number: 202103427). We recruited two CCA families (Figures 1A,B). Written informed consent was obtained from all subjects. All patients or their guardians consented to publish the clinical details. Two milliliters of blood per participant was collected and the genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, United States) for all subjects.
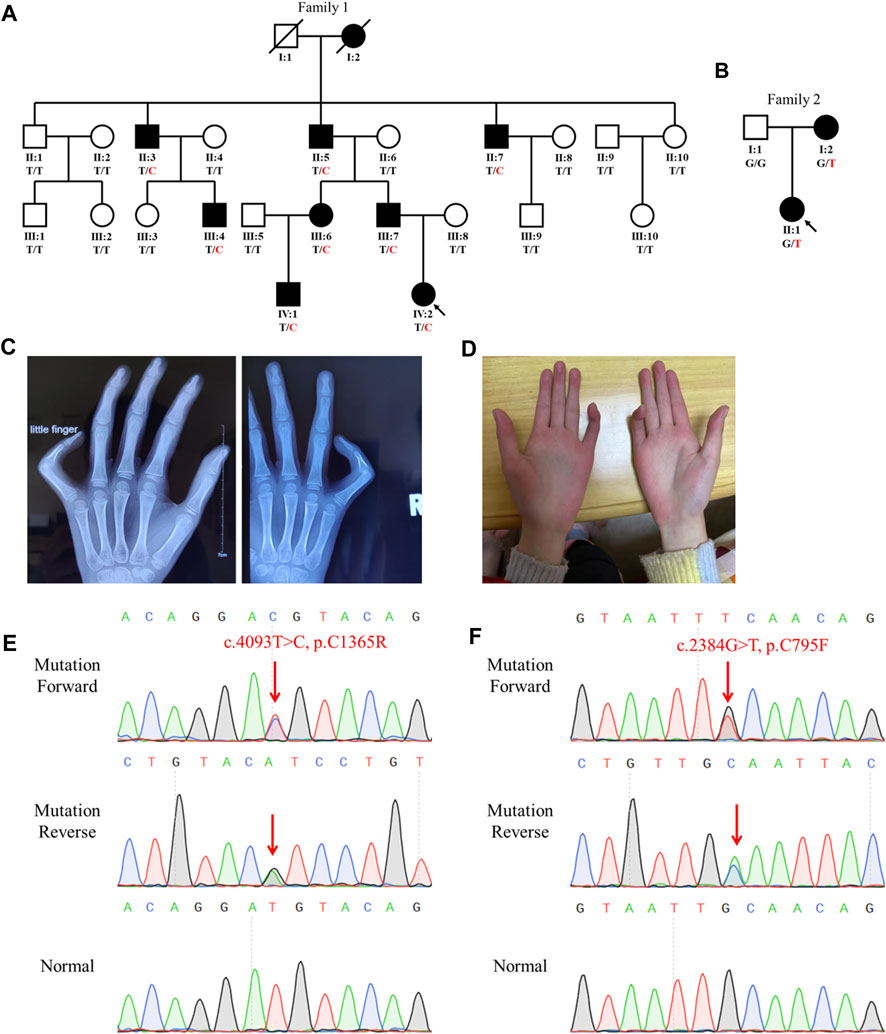
FIGURE 1. Family genogram, symptoms, and Sanger sequencing of the probands. (A,B) Genogram of the F1 family and F2 family. Black symbols represent the affected members, slashes indicate those who died, and the arrow indicates the proband. All recruited subjects underwent genetic testing, and their genotypes are identified by letters and a slash, with red representing variants. Question marks represent unknown data or symptoms. (C,D) F1 family proband had arachnodactyly and bilateral contracture of the fifth fingers. (E,F) Sequencing results of FBN2 variants using Sanger sequencing. The red arrow points to the variant site.
Whole-exome sequencingBerry Genomics Company Limited (Chengdu, China) performed the exome capture, high-throughput sequencing, and common filtering. One milligram of genomic DNA was randomly carved using a Covaris S220 sonicator (Covaris, Inc., Woburn, United States). The fragmented DNA was subjected to three enzymatic steps: end repair, A-tailing, and adapter ligation. It was amplified with Herculase II Fusion DNA Polymerase (Agilent Technologies, Inc., Santa Clara, United States). The exomes in the pre-capture libraries were captured by the SureSelect capture library kit (Agilent Technologies, Inc., Santa Clara, United States). The captured DNA library was then used for the next-generation sequencing on the Illumina HiSeq 4000 platform (Illumina Inc., San Diego, United States). Downstream processing was carried out using the Genome Analysis Toolkit (GATK), VarScan 2, and Picard, and the variant calls were made using the GATK HaplotypeCaller. Variant annotation referred to Ensembl release 82, and filtering was conducted using the ANNOVAR documentation.
We annotated the variants’ frequencies on the basis of the data from the Genome Aggregation Database (GnomAD; http://gnomad.broadinstitule.org) and 1000 Genomes Project (1000G; https://www.internationalgenome.org/) and predicted their pathogenicity using the MutationTaster (http://www.mutationtaster.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://provean.jcvi.org/index.php), and Combined Annotation–Dependent Depletion (CADD; https://cadd.gs.washington.edu/snv; variants with scores greater than 15 were considered as pathogenic). The variants were retained if they met the following two conditions: 1) the frequency of the variant was less than 0.001 and 2) at least three bioinformatics software predicted the variant to be causative. These variants were filtered when they were against the skeletal dysplasia–related genes (Supplementary Table S1). For selected variants, the inheritance patterns and clinical phenotypes of the genes were annotated using OMIM (https://www.omim.org) and the American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines was used to classify their pathogenicity grade (Richards et al., 2015).
Sanger sequencingThe FBN2 reference sequence and transcript sequence (NM_001999.3) were acquired from the NCBI (https://www.ncbi.nlm.nih.gov/gene/2201). The primer pairs used in FBN2 variant verification (FBN2 f1: 5′-AGATCTTGCCTGTGTATTTCAGTA-3′, FBN2 r1: 5′-GCTCTAAGAGATGCCAGAGAAC-3′, annealing temperature: 57.2°C; FBN2 f2: 5′-GAATTTCTGCCAGCGTCTTTC-3′, FBN2 r2: 5′-GTACCACCAAGAAGTCTGTTACT-3′, annealing temperature: 57.8°C) were designed by Integrated DNA Technologies (https://sg.idtdna.com/Primerquest/Home/Index). The polymerase chain reaction (PCR) system, which included 12.5 μL 2× PCR mix, 11 μL ultra-pure water, 0.5 μL DNA sample, 0.5 μL and 10 μM forward primer, and 0.5 μL and 10 μM reverse primer, was operated using the Biometra TOne 96G PCR amplifier (Analytik Jena, Jena, Germany). The running program was divided into four stages: 1) 95°C for 5 min; 2) sustaining 35 cycles as 95°C for 30 s, annealing temperature for 30 s, and 72°C for 1 min; 3) 72°C for 10 min; and 4) maintaining 4°C for storing the samples. The PCR products were sequenced by Sangon Biotech Company Limited (Shanghai, China).
Mutant protein modelingThe FBN2 protein structure of rats (F1M5Q4) was downloaded from the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/entry/F1M5Q4). The amino acid sequences used in the homology comparison of FBN2 were obtained from the NCBI (https://www.ncbi.nlm.nih.gov/protein/?term=FBN2). PyMOL was used to construct rat mutant FBN2 models based on the wild-type structure.
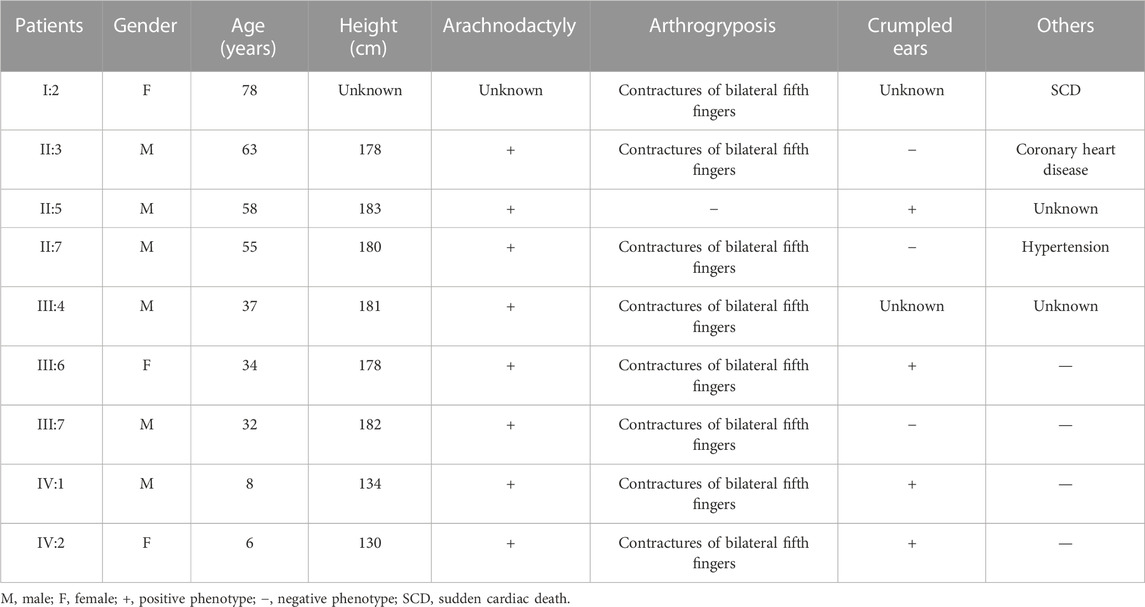
ResultsCase descriptionThe proband (IV:2) of the F1 family was a six-year-old girl from Hunan province, China (Figure 1A). She was admitted to our hospital for her contractures of bilateral fifth fingers (Figures 1C, D). She was tall (1.3 m, >0.95%) and had slender limbs, arachnodactyly, and crumpled ears (Figure 1D). Except for the abovementioned symptoms, the proband did not present other phenotypes. We thus diagnosed her with CCA. Tracing back her family history, her father (III:7) and seven family members (I:2, II:3, II:5, II:7, III:4, III:6, and IV:1) also exhibited similar phenotypes, namely, a tall and slender look, flexion contractures of proximal interphalangeal joints, and crumpled ears (Table 1). Her mother (III:8) and other subjects (II:1, II:2, II:4, II:6, II:8, II:9, II:10, III:1, III:2, III:3, III:5, III:8, III:9, and III:10) were unaffected.
TABLE 1. Phenotypes of those affected in the F1 family.
The proband (II:1) of the F2 family was a one-year-old girl. Both her mother and she exhibited arthrogryposis of bilateral third fingers and I:2 had arachnodactyly. Her father (I:1) was unaffected.
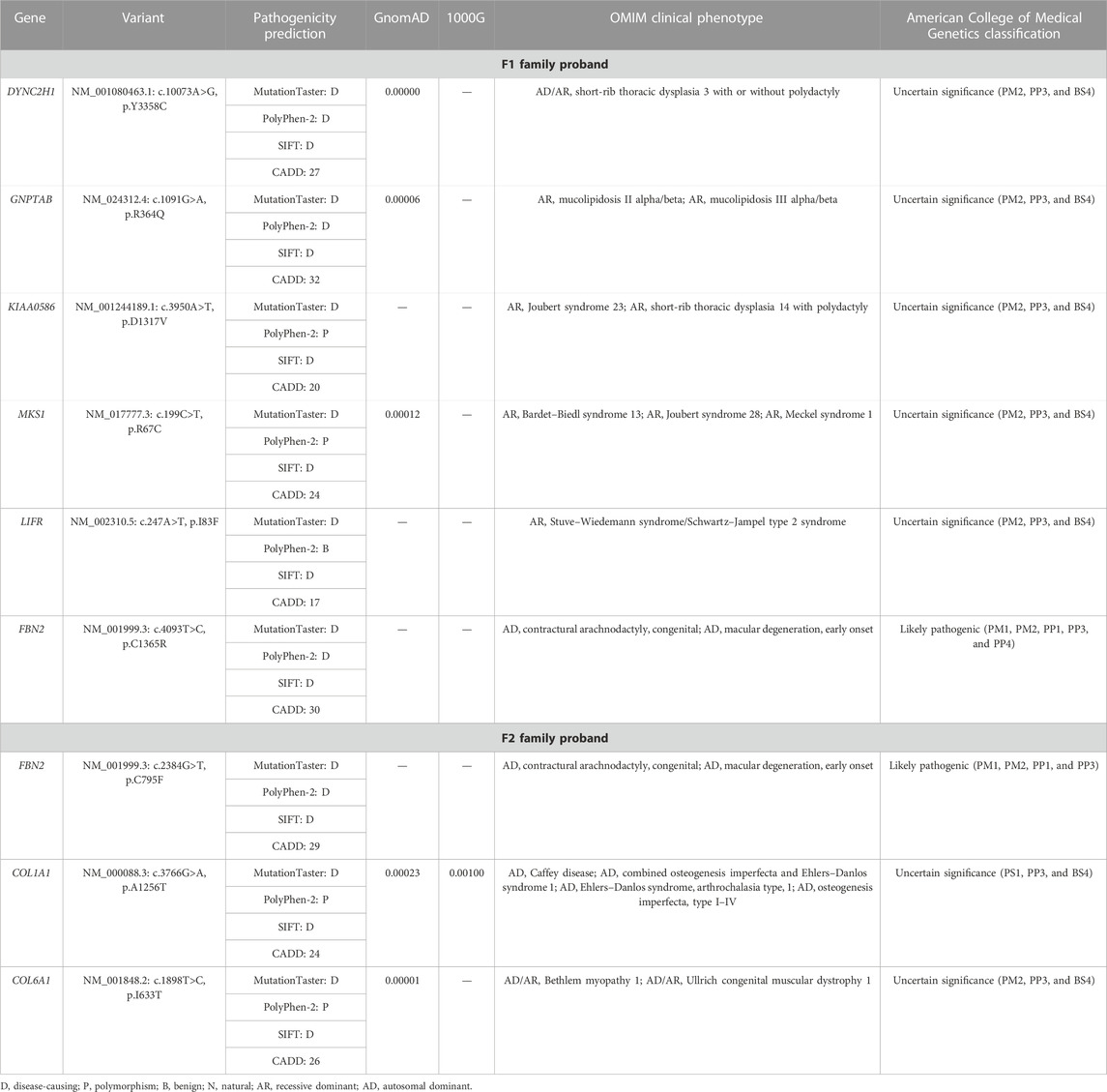
Genetic analysisWe extracted the genomic DNA of the probands’ peripheral blood and operated whole-exome sequencing (WES). We eliminated the variants with frequencies over 0.001 in GnomAD and 1000G or a prediction of benign by the MutationTaster, PolyPhen-2, SIFT, and CADD. The rest of the variants were filtered against the skeletal dysplasia–related genes (Supplementary Table S1). Finally, six variants were identified in the F1 family proband and three variants in the F2 family proband (Table 2). Adhering to the standards and guidelines of the ACMG, we classified these two FBN2 variants (NM_001999.3: c.4093T>C, p.C1365R and c.2384G>T, p.C795F) as “Likely pathogenic.”
TABLE 2. Variants identified in the two probands by WES in combination with the filtration of skeletal dysplasia–related genes.
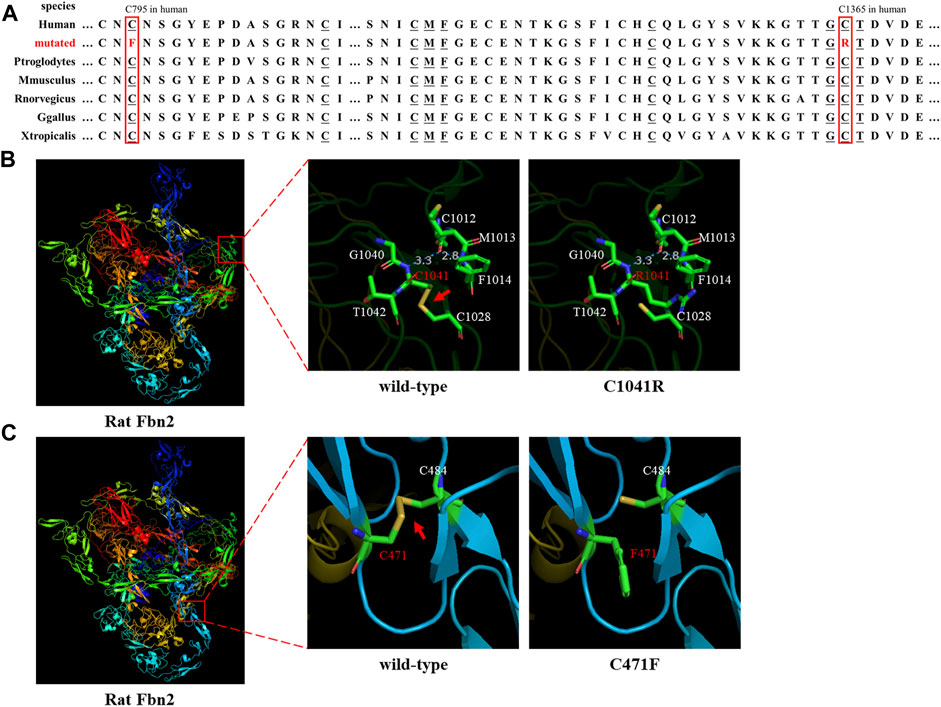
Sanger sequencing showed that the FBN2 variant c.4093T>C in the F1 proband was inherited from her father, and all patients harbored this variant (Figures 1A,E). Another FBN2 variant, c.2384G>T, was identified in the F2 proband and her mother (Figures 1B,F). The amino acid sequence alignment analysis indicated that p.C795 and p.C1365 in FBN2 were highly conserved throughout evolution (Figure 2A). According to the three-dimensional modeling of the FBN2 protein in rats, these amino acid alterations broke the disulfide bonds and may have triggered instability of the FBN2 structure, and these two regions are homologous in humans and rats (Figure 2). Thus, we reasoned that the FBN2 variants c.4093T>C, p.C1365R and c.2384G>T, p.C795F were the genetic etiologies of the F1 and F2 families, respectively.
FIGURE 2. Species conservation analysis and protein modeling. (A) Species conservation analysis of the mutant amino acid sites of FBN2. The red box represents the mutant amino acid site in various species. Underlined amino acids correspond to those labeled in Figures 2B,C that are highly evolutionarily conserved. (B,C) Three-dimensional model of rat FBN2 with wild type or p.C1041R/p.C471 variant. The red arrow points to the disulfide bond.
DiscussionPatients with CCA or MFS share many distinguishing features, such as the so-called marfanoid appearance, that is, a tall, slender, and asthenic appearance and skeletal features (Najafi et al., 2020). In contrast with MFS, most individuals with CCA have crumpled ears, flexion contractures, muscular hypoplasia, and no ocular and cardiovascular complications (Lavillaureix et al., 2017). Initially, the F1 proband was admitted to our hospital for contractures of her bilateral fifth fingers, and then we noticed her classical marfanoid appearance, that is, arachnodactyly, camptodactyly, and tall slender build (Sun et al., 2022). Given that the proband exhibited crumpled ears, without a family medical history of typical MFS-related cardiovascular diseases, we made a presumptive diagnosis of CCA. The F2 proband was a baby, and we did not do a full check and only determined her arthrogryposis.
To further determine their disorders, we performed molecular diagnosis by WES and detected a missense variant (c.4093T>C, p.C1365R) in exon 31 of FBN2 in the F1 proband. There were nine patients in this family, and Sanger sequencing showed that the variant was only identified in the affected members. In accordance with the ACMG guidelines, we determined the pathogenicity classification of the FBN2 variant as “Likely pathogenic”: 1) the variant replaced cysteine with arginine, breaking the disulfide bonds and destabilizing the protein structure (PM1); 2) the variant was absent from the controls in GnomAD and 1000G (PM2); 3) in the F1 family, only the FBN2 variant carriers presented with CCA (PP1); given that the family included nine patients across four generations and 22 living members had participated in the present study, the evidence can be considered as strong evidence of pathogenicity; 4) The MutationTaster, PolyPhen-2, SIFT, and CADD all predicted that the variant was disease-causing (PP3); and 5) the symptoms in this family suggested a suspected case of MFS or CCA and followed an autosomal dominant inheritance pattern, suggesting a monogenic disease (PP4) (Miller et al., 2021). We considered that the FBN2 variant was responsible for the disease of the F1 family and made a diagnosis of CCA in this family.
Another FBN2 variant (c.2384G>T, p.C795F) in this study that was identified in the F2 family was also classified as “Likely pathogenic” on the basis of the pathogenicity category of PM1, PM2, PP1, and PP3, as per the ACMG guidelines: 1) similar to p.C1365R, variant p.C795F also destroyed the disulfide bond, maybe destabilizing the protein structure—with many known FBN2 variants also causing the destruction of disulfide bonds (PM1); 2) the variant was not listed in the GnomAD and 1000G databases (PM2); 3) only the proband and her mother presented with symptoms in this family and harbored the variant, and the genotype and phenotype were co-segregated in this family (PP1); and 4) the bioinformatics software (MutationTaster, PolyPhen-2, SIFT, and CADD) predicted the “disease-causing” nature of this variant (PP3). The molecular diagnosis and phenotype of the mother (F2 I:2) prompted that it was a CCA family.
FBN2 contains 65 exons and codes fibrillin-2 (Kloth et al., 2021). FBN2 contains a 4-cysteine motif, 47 EGF-like domains (43 EGF-like domains with conserved calcium-binding consensus sequences called cb-EGF-like domains), two hybrid domains, seven TGF-binding proteins (TGFBP)–like modules, a glycine-rich domain, and a domain presenting with the homology to fibulins C-terminal domain III (FibuCTDIII-like motif) (Ratnapriya et al., 2014; Yin et al., 2019). In each cb-EGF-like domain, six conserved cysteine residues form three disulfide bridges for the purpose of protein stability (Davis and Summers, 2012). Missense variants can directly change the cb-EGF-like domain in the FBN2 protein and affect the constitution of the extracellular matrix microfibers. It has been shown in the following three ways: 1) variants change the Cys residue number in the cb-EGF-like domain and influence the disulfide bond formation and protein fold; 2) variants decrease the binding activity of the proteins to calcium ions and then prompt the hydrolysis of FBN2; and 3) variants affect the assembly of various domains and change the spatial structure and intermolecular interaction (Xu et al., 2020). Both the p.C1365R (cysteine changed to arginine) and p.C795F (cysteine changed to phenylalanine) variants were, respectively, located on two cb-EGF-like domains. The two altered amino acid sites were highly conserved throughout evolution (Figure 2A). Three-dimensional modeling of FBN2 in rats confirmed that the Cys residue in these motifs performed an essential role in stabilizing the structure by forming disulfide bonds, and these regions in humans were the same as those in rats (Figures 2B,C). Thus, we speculated that the FBN2 variants (c.4093T>C, p.C1365R and c.2384G>T, p.C795F) destroyed the disulfide bonds, destabilized the protein structure, affected the functions of the connective tissue, and eventually, led to the occurrence of diseases of multiple organs throughout the body.
Up till now, there have been at least 188 reported variants of FBN2 (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=FBN2). Most of these variants are associated with CCA and cluster in the cb-EGF-like domains, especially exons 24–33, which is a variant hot spot (Frederic et al., 2009). The p.C1365R variant occurred in exon 31. The missense variants (such as c.1123T>C, p.C375D; c.3769T>C, p.C1257R; c.4151G>T, p.C1384F; and c.4216T>C, p.C1406R) were identified in CCA cases and altered cysteine to other amino acids (AAs), affecting disulfide bond formation (Gupta et al., 2002; Callewaert et al., 2009; Alazami et al., 2015; Liu et al., 2015). In this study, our FBN2 variants (c.4093T>C, p.C1365R and c.2384G>T, p.C795F) also affected disulfide bond formation in the same way and led to protein misfolding, confirmed by the three-dimensional modeling of rat FBN2.
ConclusionIn summary, our study reports two novel FBN2 variants (c.4093T>C, p.C1365R and c.2384G>T, p.C795F), which were identified in two Chinese families with CCA by WES and Sanger sequencing. Based on all the factors, such as patient phenotypes, functional alterations, and in silico prediction analyses, the FBN2 variants were shown to be disease-causing and associated with CCA. Our findings have enriched the spectrum of FBN2 variants, contributing to the understanding of the correlation between genotypes and phenotypes of CCA, emphasizing the pathogenicity of disulfide bond disruption in FBN2, and serving as a reference in genetic consultation and prenatal diagnosis for CCA.
Data availability statementThe data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statementThe studies involving human participants were reviewed and approved by the Review Board of Xiangya Hospital of the Central South University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributionsA-LL and J-YJ designed the study. J-QH and RX drafted the article. LZ and J-YJ collected the samples. A-LL, J-YL, and S-HC analyzed the data and performed the experiments. RX, Y-QH, and MW edited the article. All authors read and approved the final article. All authors confirm the authenticity of the raw data.
AcknowledgmentsThe authors thank all subjects and medical personnel in this study.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, editors, and reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1035887/full#supplementary-material
ReferencesAlazami, A. M., Patel, N., Shamseldin, H. E., Anazi, S., Al-Dosari, M. S., Alzahrani, F., et al. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 10 (2), 148–161. doi:10.1016/j.celrep.2014.12.015
PubMed Abstract | CrossRef Full Text | Google Scholar
Ansari, M., Rainger, J. K., Murray, J. E., Hanson, I., Firth, H. V., Mehendale, F., et al. (2014). A syndromic form of Pierre Robin sequence is caused by 5q23 deletions encompassing FBN2 and PHAX. Eur. J. Med. Genet. 57 (10), 587–595. doi:10.1016/j.ejmg.2014.08.007
PubMed Abstract | CrossRef Full Text | Google Scholar
Buchan, J. G., Alvarado, D. M., Haller, G. E., Cruchaga, C., Harms, M. B., Zhang, T., et al. (2014). Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum. Mol. Genet. 23 (19), 5271–5282. doi:10.1093/hmg/ddu224
PubMed Abstract | CrossRef Full Text | Google Scholar
Callewaert, B. (1993). “Congenital contractural arachnodactyly,” in GeneReviews. Editors M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (Seattle: University of Washington).
Callewaert, B. L., Loeys, B. L., Ficcadenti, A., Vermeer, S., Landgren, M., Kroes, H. Y., et al. (2009). Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: Report of 14 novel mutations and review of the literature. Hum. Mutat. 30 (3), 334–341. doi:10.1002/humu.20854
PubMed Abstract | CrossRef Full Text | Google Scholar
Davis, M. R., and Summers, K. M. (2012). Structure and function of the mammalian fibrillin gene family: Implications for human connective tissue diseases. Mol. Genet. Metab. 107 (4), 635–647. doi:10.1016/j.ymgme.2012.07.023
PubMed Abstract | CrossRef Full Text | Google Scholar
Du, Q., Zhang, D., Zhuang, Y., Xia, Q., Wen, T., and Jia, H. (2021). The molecular genetics of marfan syndrome. Int. J. Med. Sci. 18 (13), 2752–2766. doi:10.7150/ijms.60685
PubMed Abstract | CrossRef Full Text | Google Scholar
Frederic, M. Y., Monino, C., Marschall, C., Hamroun, D., Faivre, L., Jondeau, G., et al. (2009). The FBN2 gene: New mutations, locus-specific database (universal mutation database FBN2), and genotype-phenotype correlations. Hum. Mutat. 30 (2), 181–190. doi:10.1002/humu.20794
PubMed Abstract | CrossRef Full Text | Google Scholar
Gupta, P. A., Putnam, E. A., Carmical, S. G., Kaitila, I., Steinmann, B., Child, A., et al. (2002). Ten novel FBN2 mutations in congenital contractural arachnodactyly: Delineation of the molecular pathogenesis and clinical phenotype. Hum. Mutat. 19 (1), 39–48. doi:10.1002/humu.10017
PubMed Abstract | CrossRef Full Text | Google Scholar
Kloth, K., Neu, A., Rau, I., Hulsemann, W., Kutsche, K., and Volk, A. E. (2021). Severe congenital contractural arachnodactyly caused by biallelic pathogenic variants in FBN2. Eur. J. Med. Genet. 64 (3), 104161. doi:10.1016/j.ejmg.2021.104161
PubMed Abstract | CrossRef Full Text | Google Scholar
Lavillaureix, A., Heide, S., Chantot-Bastaraud, S., Marey, I., Keren, B., Grigorescu, R., et al. (2017). Mosaic intragenic deletion of FBN2 and severe congenital contractural arachnodactyly. Clin. Genet. 92 (5), 556–558. doi:10.1111/cge.13062
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, W., Zhao, N., Li, X. F., Wang, H., Sui, Y., Lu, Y. P., et al. (2015). A novel FBN2 mutation in a Chinese family with congenital contractural arachnodactyly. FEBS Open Bio 5, 163–166. doi:10.1016/j.fob.2015.02.005
PubMed Abstract | CrossRef Full Text | Google Scholar
Miller, D. T., Lee, K., Chung, W. K., Gordon, A. S., Herman, G. E., Klein, T. E., et al. (2021). ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of medical genetics and genomics (ACMG). Genet. Med. 23 (8), 1381–1390. doi:10.1038/s41436-021-01172-3
PubMed Abstract | CrossRef Full Text | Google Scholar
Najafi, A., Caspar, S. M., Meienberg, J., Rohrbach, M., Steinmann, B., and Matyas, G. (2020). Variant filtering, digenic variants, and other challenges in clinical sequencing: A lesson from fibrillinopathies. Clin. Genet. 97 (2), 235–245. doi:10.1111/cge.13640
PubMed Abstract | CrossRef Full Text | Google Scholar
Peeters, S., Decramer, A., Cain, S. A., Houpt, P., Verstreken, F., Noyez, J., et al. (2021). Delineation of a new fibrillin-2-opathy with evidence for a role of FBN2 in the pathogenesis of carpal tunnel syndrome. J. Med. Genet. 58 (11), 778–782. doi:10.1136/jmedgenet-2020-107085
PubMed Abstract | CrossRef Full Text | Google Scholar
Qiu, J., Lou, Y., Zhu, Y., Wang, M., Peng, H., Hao, Y., et al. (2021). Clinical characteristics and genetic analysis of a family with birt-hogg-dube syndrome and congenital contractural arachnodactyly. Front. Genet. 12, 768342. doi:10.3389/fgene.2021.768342
PubMed Abstract | CrossRef Full Text | Google Scholar
Ratnapriya, R., Zhan, X., Fariss, R. N., Branham, K. E., Zipprer, D., Chakarova, C. F., et al. (2014). Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Hum. Mol. Genet. 23 (21), 5827–5837. doi:10.1093/hmg/ddu276
PubMed Abstract | CrossRef Full Text | Google Scholar
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
PubMed Abstract | CrossRef Full Text | Google Scholar
Sun, L., Huang, Y., Zhao, S., Zhong, W., Shi, J., Guo, Y., et al. (2022). Identification of novel FBN2 variants in a cohort of congenital contractural arachnodactyly. Front. Genet. 13, 804202. doi:10.3389/fgene.2022.804202
PubMed Abstract | CrossRef Full Text | Google Scholar
Xu, P., Li, R., Huang, S., Sun, M., Liu, J., Niu, Y., et al. (2020). A novel splicing mutation in the FBN2 gene in a family with congenital contractural arachnodactyly. Front. Genet. 11, 143. doi:10.3389/fgene.2020.00143
PubMed Abstract | CrossRef Full Text | Google Scholar
Yin, W., Kim, H. T., Wang, S., Gunawan, F., Li, R., Buettner, C., et al. (2019). Fibrillin-2 is a key mediator of smooth muscle extracellular matrix homeostasis during mouse tracheal tubulogenesis. Eur. Respir. J. 53 (3), 1800840. doi:10.1183/13993003.00840-2018
留言 (0)