
記住我




This was a Phase I multicentre randomized, open, parallel-arm clinical trial with the investigational medicinal product (IMP) [111In]In-CP04 ([111In]In-DOTA-(DGlu)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2), registered at www.clinicaltrials.gov (NCT03246659) and EudraCT (2015-000805-38). The trial was approved by the ethics committees in all three centres participating in the clinical part of the project. The Investigator’s Brochure (IB) and the Investigational Medicinal Product Dossier (IMPD) were submitted and approved by the national authorities of all participating centres.
Study populationPatients meeting the inclusion criteria (Table 1) were enrolled after providing a signed informed consent for trial participation. Women of childbearing age and fertile men were asked to apply effective methods of contraception before and after enrolment throughout the study up to the last follow-up visit. If applicable, a pregnancy test was performed at the screening visit before enrolment in the study, on the day of radiopharmaceutical administration(s) and at the last follow-up visit. The assessment of baseline laboratory (complete blood count, biochemistry) and tumour-related signs and symptoms was performed before the start of the clinical trial, with appropriate follow-up.
Table 1 Inclusion and exclusion criteria for the clinical trialStudy objectives and endpointsThe primary objective of the trial was the evaluation of the safety of the administration of [111In]In-CP04 at low mass amount of 10 μg CP04 and at high mass amount of 50 μg CP04 radiolabelled with 200±10% MBq of 111In. Therefore, the definition of the type, frequency, severity, timing, and relation to the studied radiopharmaceutical administration of Adverse Events assessed by CTCAE v4.0 was used as primary endpoint to define the safety and tolerability of [111In]In-CP04.
The secondary objectives of the study include the evaluation of dosimetry and biodistribution of the IMP with the corresponding endpoints being the determination of the (i) kinetic curve and biodistribution profile by assessment of [111In]In-CP04 uptake in normal organs, tumour, and metastases; (ii) effective dose equivalent (mSv) and absorbed doses (mGy) of normal organs; (iii) effect of the IMP administered mass amount on the biodistribution and nephroprotection (detailed in the “Study design” section); (iv) pharmacokinetics of [111In]In-CP04, metabolism and excretion based on blood and urine sampling analysis. Furthermore, the detection rate of [111In]In-CP04 scintigraphy compared to other diagnostic tests ( [18F]FDG PET/CT, ceCT, or MRI imaging) was used as endpoint to determine the diagnostic performance of [111In]In-CP04 scan.
Study designThe clinical trial design comprised of two phases, 1A and 1B. In Phase 1A, the first 4 enrolled patients (3 patients with low-burden advanced disease and one with negative imaging and elevated Ct) were administered with the low mass amount of 10 μg [111In]In-CP04 as a safety step. If this dose was well tolerated, the high mass amount of 50 μg of [111In]In-CP04 was injected after 2 weeks. The mass amount of 50 μg was assumed to be the potential therapeutic amount if the compound was to be labelled with the β-emitter lutetium-177 (177Lu). According to the study protocol, subsequent tracer administrations were performed at intervals of at least 2 weeks, providing that no serious adverse events (SEA) were observed (Fig. 1) [9]. After safety confirmation, in Phase 1B only 50 μg of [111In]In-CP04 was administered to all further enrolled patients. Additionally, these patients were randomly assigned into Arm 1 with Gelofusine and into Arm 2 without Gelofusine co-administration, respectively.
Fig. 1
Detailed physical examinations with vital sign assessment, 12-lead ECG, and laboratory testing were performed to investigate the safety of intravenous administration of [111In]In-CP04. The overall safety profile of the tested CCK-2/gastrin receptor ligand as characterized by timing, type, frequency, severity of adverse events (AE), and laboratory abnormalities was based on Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Since this was a Phase I clinical trial, the project was assumed as having a high-risk potential for the patient, and therefore, special care was taken with the monitoring of the study. In total, eight control visits were carried out over 4 months of the follow-up with physical examination, laboratory testing, and ECG. Additionally, Ct and proCt concentrations were measured after each [111In]In-CP04 injection at predefined time points (0, 2, 5, 10, 20 min).
[111In]In-CP04 preparationWithin the preclinical phase of GRAN-T-MTC, [111In]In-CP04 was evaluated for pharmaceutical characteristics and validated in animal models. A clinically useful kit formulation for CP04 radiolabelling with 111In for use in the clinical phase has been developed [10]. The results of the preclinical part of the project, as well as the protocol of the clinical study, have been already published elsewhere [9, 11].
The cold CP04 kits with mass amounts of 10 and 50 μg for on-site radiolabelling were used. Radiolabelling with 111In (Indium chloride, Mallinckrodt Medical, Petten) was performed at 90°C using a standardized protocol resulting in a final preparation containing 200±10% MBq [111In]In-CP04 ready for injection [11]. The radiolabelling procedure was performed in an environment suitable for handling radioactive material at the Radiopharmacy Department or at the site specially designated by the Nuclear Medicine Department in each participating centre. Quality control by high-performance liquid chromatography (HPLC) ensured a radiochemical purity of [111In]In-CP04 of >95% for all applications.
Prior to any patient administration, the Standard Operating Procedures (SOP) were developed for radiolabelling and quality control procedures, and the personnel of each clinical centre was trained and qualified on these SOP for their implementation in accordance with the IMPD.
Gamma camera calibration and validation for quantitative SPECT imagingPrior to the start of the clinical trial, a cross-calibration of all SPECT/CT cameras and the dose calibrators in the participating centres was performed using a Jaszczak SPECT phantom to ensure the accuracy and reproducibility of calculations. An empty Jaszczak phantom with all inserts removed was filled with a homogenous 111In solution (50 MBq). The acquisition and reconstruction protocol for the phantom were identical to the acquisition settings for the 24 h post-injection quantitative SPECT/CT scan conducted during the trial. These procedures were standardized across sites according to the study protocol and were carried out under supervision of an experienced study dosimetrist or physicist.
Radiopharmaceutical administration and imagingThe duration of [111In]In-CP04 administration with the low and high mass amount was 5 min and 30 min, respectively. During the injection of the IMP, a dynamic scan of the kidneys and stomach was performed for 30 min (anterior and posterior acquisition). The imaging protocol included a mandatory whole-body scan (WBS) at 1 h, 4 h, 24 h, and 48 h p.i. (table shift speed 12 cm/min). A quantitative SPECT/CT scan of the abdomen was performed at 24 h p.i. as well as an additional non-quantitative SPECT/CT scan of the area where pathological lesions were detected during WBS, if the scan duration was tolerated by the patient. Gelofusine (B. Braun, Melsungen, Germany) was administered as an infusion of 1 mL/kg body weight over 10 min followed by a 3-h infusion of 0.02 mL/kg/min after the injection of the IMPD. Physiological parameters (heart rate, blood pressure, oxygen saturation) were repeatedly measured during and after the [111In]In-CP04 infusion as per study protocol.
Biodistribution and dosimetry evaluationDuring the [111In]In-CP04 administration blood samples for dosimetry and pharmacokinetic purposes were taken at the set time intervals according to the study protocol: biodistribution and dosimetry data were acquired based on serial planar and SPECT/CT images, and blood and urine collection at regular time intervals. Urine samples were collected up to 24 h after the tracer injection to determine the bladder wall absorbed dose. On the WBS planar imaging at 1, 4, 24, and 48 h (including standard activity) and quantitative SPECT/CT imaging at 4 and 24 h, the regions of interest (ROI) were drawn over the kidneys and over the lesions when visible. The counts in the ROIs were quantified to activity by cross-calibration with the activity determined in the corresponding volumes of interest (VOIs) drawn on the quantitative SPECT/CT image at 24 h. Time-activity curves were determined by fitting exponential curves through the uptake data. Time integrated activity coefficients (TIAC) were calculated for 111In and 177Lu by integration of the TIACs multiplied with their physical decaty functions. The absorbed dose per unit activity in each organ was determined by multiplication with the 111In and 177Lu S-values according to the MIRD formalism. The blood time-activity curve was used to estimate the absorbed dose to the bone marrow. The activity concentration in the bone marrow was considered to be equal to the blood activity concentration. The activity and volumes of the urine samples were used to determine the urinary clearance rate, and with the dynamic bladder model, the TIAC in the bladder and the absorbed dose to the bladder wall were determined. The effect on the kidney absorbed dose after co-administration of the nephroprotective agent Gelofusine was evaluated. All dosimetry calculations were performed with the Hermes-OLINDA v2.1 hybrid 2D dosimetry software.
Ct and proCt measurementsDuring the [111In]In-CP04 administration blood samples for Ct and proCt concentrations measurements were taken at predefined time intervals according to the study protocol. The measurements were performed at the Central Laboratory participating in the external international quality assessment scheme, served by RANDOX Laboratories Ltd., Crumlin, UK. The enzyme-linked immunosorbent assay (ELISA) with the Calcitonin EIA-3648 and Procalcitonin (Human) EIA-5291reagent kits (DRG®Instruments GmbH,Germany) and ELx808 spectrophotometer (BioTek R Instruments, Inc., Winooski, Vermont, United States) was used. The obtained blood samples were centrifuged (15 min, 1000 g, 18°C) and the serum after separation from the clot was stored at −80°C locally at each centre. Serum samples were sent frozen in dry ice to the Central Laboratory. For each shipment, a temperature data logger was used to monitor and record temperature values during the whole period.
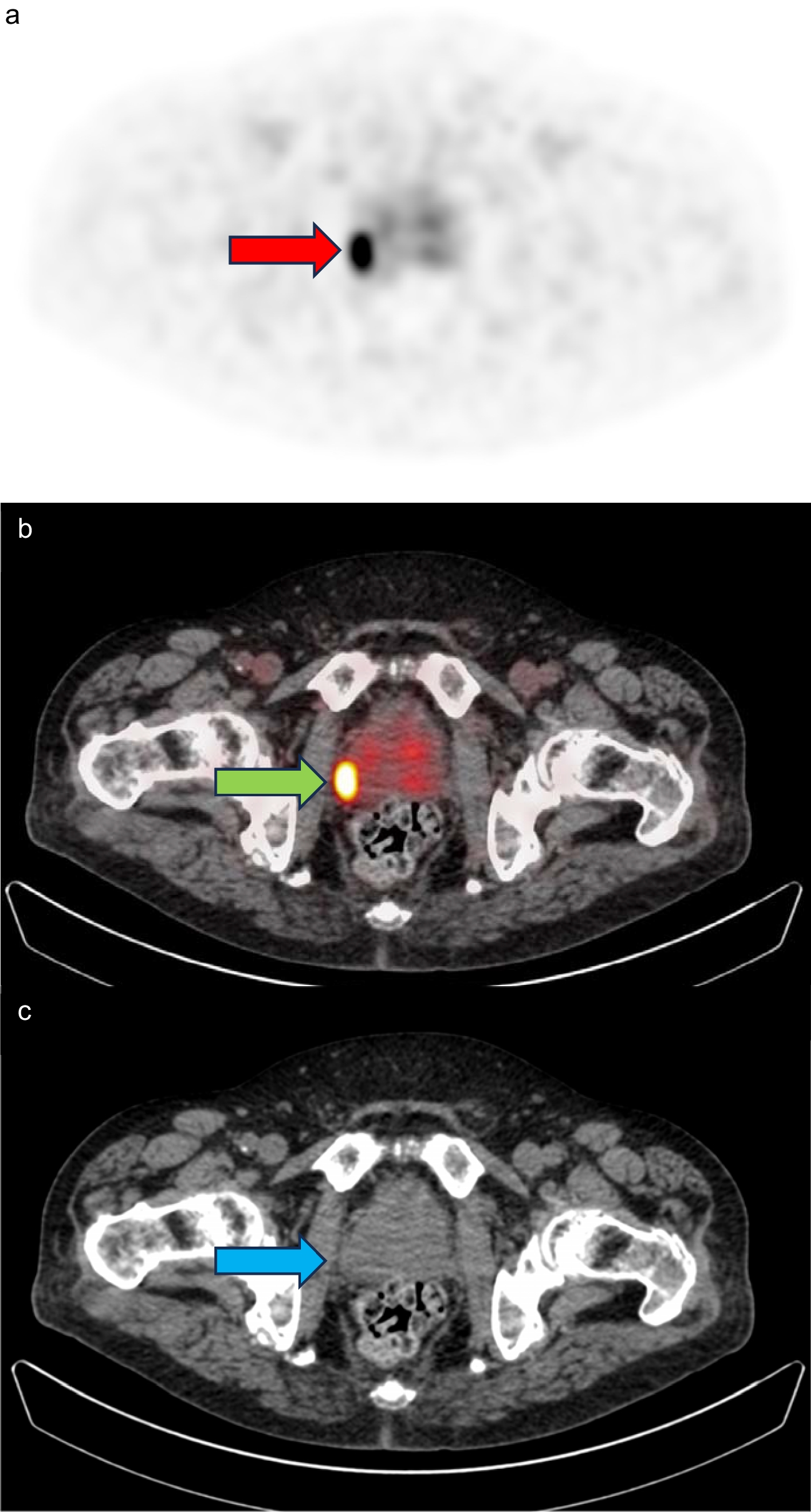
Assessment of the diagnostic potential of [111In]In-CP04For the evaluation of the acquired images, qualitative visual analysis was used to determine the presence/absence and the number of lesions identified by scintigraphy based on abnormal tracer uptake (defined as clearly discernible uptake above background concentration—regional blood pool). The number and identity of the lesions with increased abnormal uptake detected per verifiable organ or body region relative to those detected by conventional imaging were assessed. Identification of the time points p.i. with the highest observed number of lesions and the highest tumour/background ratios for both 10 and 50 μg mass amounts was performed to evaluate the influence of mass amount on tumour and normal organ uptake. In all clinical centres, the scintigraphy scans were evaluated by two experienced nuclear medicine specialists.
Statistical analysisThe assessed variables were analyzed and shown descriptively. T-test (paired/unpaired, as appropriate) was used for comparison of variables. All statistical calculations were performed using SPSS and STATA software packages.
留言 (0)