
記住我
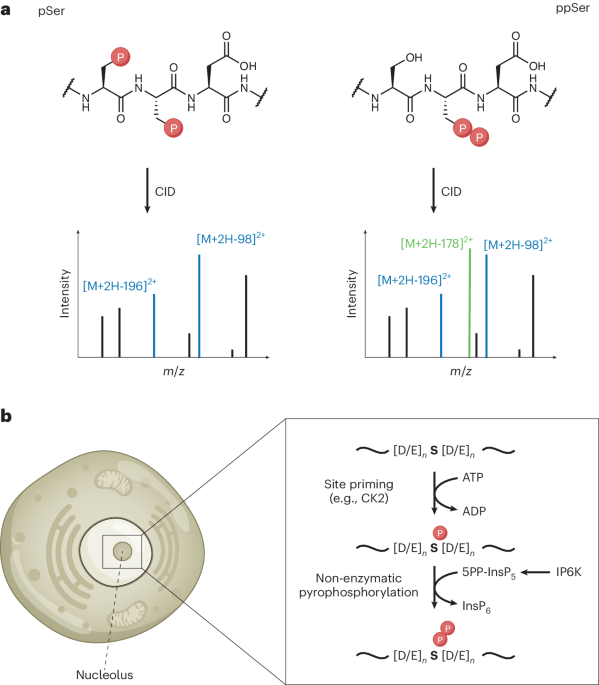

Detailed procedures for the synthesis of the probes are given in the Supplementary Note.
Molecular biologyA pET51b(+) vector (Novagen) was used for protein production in E. coli. Proteins were N-terminally tagged with Strep-tag and C-terminally tagged with Hisx10, respectively. A pcDNA5/FRT/TO vector (Thermo Fisher Scientific) was used for generating HEK293 cells that stably express sensor proteins.
The plasmids encoding the genes of PanK1, PanK2, PanK3, COASY, SLC25A16, and SLC25A42 were obtained from Addgene plasmid repository (Supplementary Data). The genes of PPCS, PPCDC, DCAKD, ACLY, and NUDT8 were amplified from cDNA, which was prepared by QuantiTect Reverse Transcription Kit (Qiagen) using total RNA of HEK293 cells. The genes of E. coli PANK were synthesized by Eurofins Genomics. These genes were subsequently cloned into PET51b(+) or pcDNA5/FRT vector by Gibson Assembly.
Protein production and purificationThe ecPanK variants, human PanKs (hPanK1, hPanK2 and hPanK3), and sensor proteins were expressed in E. coli strain BL21(DE3). The bacterial culture was incubated at 37 °C to reach an optical density at 600 nm (OD600) of 0.8. The protein expression was induced by adding 1.0 mM isopropyl β-d-thiogalactopyranoside (IPTG) to the culture. The culture was then cooled to 16 °C and incubated overnight while shaking at 220 r.p.m. After 20 h, the bacteria were harvested by centrifugation at 4,000g for 10 min and lysed by sonication in the presence of 1 mg ml−1 lysozyme and 1 mM phenylmethylsulfonyl fluoride (PMSF). The cell lysate was cleared by centrifugation at 20,000g and 4 °C for 20 min. All proteins were purified using Ni-NTA affinity chromatography (Qiagen), which was followed by Strep-Tactin purification (IBA Lifesciences), according to the manufacturer’s protocol. The protein concentration was determined by measuring the absorbance at 485 nm (εsfGFP = 83,300 M−1cm−1). The purified proteins were diluted in phosphate-buffered saline (PBS) (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) to 50 μM, aliquoted, flash frozen in liquid nitrogen and stored at −80 °C until use.
Engineering of ecPanKIn a fluorescence polarization competition assay with TMR-TAZ, ecPanK only exhibited a 6.6-fold higher affinity for CoA over acetyl-CoA (AcCoA) (Extended Data Fig. 1e) and we thus attempted to increase the specificity of ecPanK for CoA over AcCoA. The sulfhydryl moiety (-SH) can interact favorably with aromatic rings through thiol-aromatic interactions51, and we therefore mutated L277 in the binding pocket of ecPanK to W (Fig. 2b). We furthermore introduced the mutation F252Y to sterically discriminate against AcCoA. The resulting double mutant L277W, F252Y displayed a 21-fold higher affinity for CoA over AcCoA (Supplementary Table 1). Furthermore, the catalytic activity of ecPanK was abolished by implementing the D127A mutation in the active site52 (Extended Data Fig. 2g). The triple mutant ecPanKD127A,F252Y,L277W exhibited a Kd value of 21.6 ± 3.7 μM for TMR-TAZ whereby CoA competed with TMR-TAZ for binding (Supplementary Table. 1). For the circular permutation of ecPanK, we introduced new termini near the substrate binding site at residues D213 and P214 and connected the original termini by a flexible linker GSGGTG, resulting in cpecPanKD127A,F252Y,L277W, which was used for the generation of CoA-SnifitG41. The point mutations were made using the Q5 site directed mutagenesis kit according to the manufacturer’s protocol.
HaloTag labeling in vitroThe sensor proteins (1 μM) were labeled in PBS spiking labeling substrate 8 iterative times every 15 min to reach a final concentration of 4 μM. After that, the mixture was incubated at room temperature for another 1 h, the excess of probe was washed out (two cycles) using a centrifugal filter device (Microcon YM-50, Millipore) using PBS. The labeled sensors were stored at 4 °C until further use.
In vitro PanK activity assaysThe activity of wild-type ecPanKWT, the dead mutant ecPanKD127A and hPanKs was determined by performing absorbance kinetics on a microplate reader (Spark 20 M, Tecan). The production of ADP was coupled to the consumption of NADH, by using pyruvate kinase (PK, Sigma-Aldrich) and lactate dehydrogenase (LDH, Sigma-Aldrich). The reaction mix (200 μl) contained 50 mM Tris-HCl, 10 mM MgCl2, 20 mM KCl, 1.5 mM ATP, 0.5 mM NADH, 0.5 mM phosphoenolpyruvate, 3 units of PK, 3 units of LDH, 0.5 μM of PanK, pH 7.6. The reaction was initiated by the addition of 200 μM pantothenate and was monitored by following the decrease in absorption at 340 nm at 25 °C.
Sensor titrationOne hundred nanomolar sensors were titrated in PBS, supplemented with 50 mM HEPES and 0.5 mg ml−1 bovine serum albumin (BSA) with defined concentrations of CoA in a final volume of 50 μl in black flat-bottom non-binding 96-well plates (FALCON). After 5 min incubation at 25 °C, fluorescence emission spectra were recorded on a microplate reader (Spark 20M, Tecan). The sensor was excited at 450 nm (bandwidth 10 nm) and the emission spectra were recorded from 480 to 650 nm (bandwidth 10 nm) with a step size of 2 nm. FRET ratios (sfGFP/MaP) were calculated from the emission intensity of sfGFP (510 nm) and MaP dye (580 nm), and further plotted against the CoA concentration. To obtain the concentration of half-maximal ratio change, c50, the equation (1) was fitted to the data:
$$R = R_} + \frac} - R_}}}}}]}}}}$$
(1)
Where R is the experimental FRET ratio, [CoA] is the concentration of free CoA, and Rmin and Rmax are the FRET ratio in absence and at saturation of CoA, respectively. Fits were performed using OriginPro 2021 with free fit parameters c50, Rmin, and Rmax. Dynamic range (maximum ratio change of emission intensity) was calculated as ΔR = Rmax/Rmin.
Fluorescence polarizationFifty nanomolar TMR-TAZ was incubated with varying amounts of PanK proteins for 5 min at room temperature in PBS supplemented with 0.5 mg ml−1 BSA. Assays were performed in non-binding black 96-well plates (Corning) with a final volume of 50 μl and were measured on a microplate reader (Spark 20M, Tecan) with excitation at 520 nm (bandwidth 20 nm) and emission 600 nm (bandwidth 20 nm). Fluorescence polarization (FP) was calculated according to equation (2).
$$\mathrm = \frac}}$$
(2)
Where FP is the fluorescence polarization, I∥ is the fluorescence intensity parallel to the excitation light polarization, I⊥ is the fluorescence intensity perpendicular to the excitation light polarization, and G is the grating factor (G = I∥/I⊥). Three independent titrations were performed for each protein variant. The equation (3) was fitted to the data using OriginPro 2021.
$$\begin\mathrm = \mathrm_ + \left( _ - _}} \right)\\ \times \frac + \left[ } \right]} \right) - \left. } \right] + K_\mathrm} \right)^2 - 4\left[ L \right]\left[ } \right]} } \right)}}}\end$$
(3)
Where FP0 is the fluorescence polarization of the free fluorophore, FPs is the fluorescence polarization of the fluorophore bound with protein, Kd is the dissociation constant, [protein] is the concentration of ecPanK proteins, and [L] is the concentration of TMR-TAZ, which was 50 nM in this case.
The binding affinity (c50) of different ecPanKs for CoA and AcCoA were determined by a fluorescence polarization competition assay against TMR-TAZ. 50 nM TMR-TAZ and 20 μM ecPanKs, except ecPanKD127A,F252Y,L277W (100 μM), were titrated against CoA or AcCoA concentrations ranging from 64 nM to 5 mM in PBS supplemented with 0.5 mg ml−1 BSA and 50 mM HEPES. Assays were performed in non-binding black 96-well plates with a final volume of 50 μl with excitation at 520 nm (bandwidth 20 nm) and emission 600 nm (bandwidth 20 nm). Obtained FP values were averaged and fitted to equation (4) to estimate the c50 values using OriginPro 2021.
$$\mathrm = \mathrm_ + \frac_ - _}}}}}}}}$$
(4)
Where FP0 is the fluorescence polarization of the fluorophore with ecPanKs in the absence of CoA or AcCoA (bound fluorophore), FPs is the fluorescence polarization of the fluorophore in the presence of CoA or AcCoA (free fluorophore) c50 is the half-maximal effective concentration, and [L] is the concentration of CoA or AcCoA.
Cell cultureHEK293 cells, which refers to the Flp-In-T-REx-293 cell lines (Thermo Fisher Scientific) in this study, HeLa, and U-2 OS cell lines were grown in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX-I, 1 mM pyruvate (Gibco) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Gibco), and HepG2 cell line was grown in RPMI-1640 medium (Gibco) supplemented with 10% (v/v) heat-inactivated FBS. All cells were grown at 37 °C, 5% CO2. Cells stably expressing the sensor protein were generated according to the standard protocol from Thermo Fisher Scientific. In brief, Flp-In-T-REx-293 cells were co-transfected with pOG44 and pcDNA5/FRT/TO plasmid encoding Flp-recombinase and sensor proteins, respectively, followed by selection with hygromycin B (100 μg ml−1). To induce protein expression, cells expressing cytosolic sensors were incubated in the presence of 400 ng ml−1 doxycycline for 12 h and cells expressing mitochondrial sensors were incubated in the presence of 4 ng ml−1 doxycycline for 12 h.
Subcellular colocalizationThe subcellular localization of cytosolic and mitochondrial sensor was determined in living cells by staining the cells with commercial fluorescent probes (see below). The localization of PanKs, Nudt8 and CG4241 was determined by transiently transfection of C-terminal GFP-tag fusion proteins (Extended Data Fig. 8). We assume that the localization of PanKs, SLC25A16, SLC25A42, and CG4241 is not affected by their overexpression, as we could not verify their localization through immunostaining with commercial antibodies.
For the cytosolic and mitochondrial sensors, cells stably expressing the sensors were seeded in a glass-bottom 96-well cell imaging plate and the sensor expression was induced in full growth medium at 37 °C, 5% CO2 for 12 h with 400 and 4 ng ml−1 doxycycline, respectively. For PanK1, PanK2, PanK3, and CG4241, HEK293 transiently transfected with PanK1–GFP (catalytic core of PanK1α), PanK2–GFP, PanK3–GFP, and CG4241–GFP fusion proteins were allowed to grow for 24 h after transfection.
The cells were labeled in full growth medium with 100 nM MitoTracker Red CMXRos and 1 μg ml−1 Hoechst 33342 at 37 °C, 5% CO2 for 1 h. Excess of dye was removed by washing twice with HBSS supplemented with 0.2 mg ml−1 BSA. The medium was exchanged with HBSS before imaging. The images were analyzed with ImageJ53.
ImmunostainingThe HEK293 cells transiently transfected with pcDNA5/FRT plasmid encoding NUDT8 gene were grown on a glass-bottom 96-well cell imaging plate for 24 h. After staining with 100 nM MitoTracker Red CMXRos for 1 h, the cells were fixed with 4% PFA in PBS for 15 min at 25 °C, followed by permeabilization with 0.3% Triton X-100 in PBS. The cells were washed with PBS and blocked with 3% BSA in PBS for 1 h at 25 °C and then incubated for 12 h at 4 °C with 1% BSA in PBS containing 2 μg ml−1 rabbit primary polyclonal antibody Nudt8 (PA5-59493, 1:50, Thermo Fisher Scientific), followed by 1 h incubation with Alexa Fluor 647 goat anti-rabbit IgG antibody (A32728, 1:1,000, Invitrogen) and 1.0 μg ml−1 Hoechst 33342 at 25 °C. The cells were washed three times with PBS before imaging.
Capillary immunoblotting analysisImmunoblotting was performed using the system Wes (ProteinSimple) according to the user manual. The cells were extracted using Cell Lysis Reagent (Sigma-Aldrich). Total lysate was mixed with a master mix (ProteinSimple) to a final concentration of 1× sample buffer, 1× fluorescent molecular weight marker and 40 mM dithiothreitol, then heated at 95 °C for 5 min. The samples, blocking reagent, primary antibodies, horse-radish-peroxidase-conjugated secondary antibody, chemiluminescent substrate, separation and stacking matrices were also dispensed to designated wells in 25 well plates. After plate loading, separation electrophoresis and immunodetection steps took place in the capillary system and were fully automated. Capillary immunoblotting analysis was carried out at room temperature, and instrument default settings were used. Capillaries were first filled with separation matrix followed by stacking matrix, and about 40 nl sample loading. During electrophoresis, proteins were separated on the basis of molecular weight through the stacking and separation matrices at 250 V for 40 min and then immobilized on the capillary wall using proprietary photo-activated capture chemistry. The matrices were then washed out. Capillaries were next incubated with a blocking reagent for 15 min, and target proteins were immunoprobed with primary antibodies to GAPDH (NB300-322, 1:1,000, Novus Biologicals), Nudt8 (PA5-59493, 1:50, Thermo Fisher Scientific), PanK2 (PA5-52563, 1:20, Thermo Fisher Scientific), COASY (WH0080347M1-100UG, 1:500, Sigma-Aldrich), and ACLY (PA5-29497, 1:100, Thermo Fisher Scientific), followed by anti-rabbit detection module (DM-001, ProteinSimple) or anti-mouse detection module (DM-002, ProteinSimple) secondary antibodies, addition of chemiluminescence detection mixture (ProteinSimple) and imaging. The images were analyzed by Compass for SW version 4.0.0 (ProteinSimple).
Labeling sensor protein in mammalian cellsAfter induction with doxycycline, the cells stably expressing sensor proteins were labeled with 1 μM fluorescent probe (Halo-MaP-TAZ, 1 mM stock in dimethy sulfoxide (DMSO), 1000×) in fresh pre-warmed full growth medium supplemented with 10% FBS at 37 °C, 5% CO2 for 12 h. Then, the cells were washed once with HBSS supplemented with 0.2 mg ml−1 BSA and incubated in this buffer before imaging.
Confocal microscopyImages of HEK293 cells labeled with probes were taken using a Leica TCP SP8 confocal microscope equipped with a 40× plan Apochromat 1.4 numerical-aperture water-immersion objective lens. As excitation source, the white light laser was set to 480 nm with 80 MHz pulse frequency for GFP excitation and was set to 535 nm with 80 MHz pulse frequency for MaP dye excitation. Fluorescence signal was collected at 490–540 nm for GFP and at 560–620 nm for MaP, respectively. The scanning parameters were set to 1.5× zoom, scan speed 200 Hz, pixel size 0.379 μm, image format 512 × 512 pixels, pinhole 77 μm. Unlabeled cells were used as the donor-only sample. The HEK293 cells stably expressing HaloTag-(EAAAK)5-ecPanK fusion, on which sfGFP was removed, were labeled with Halo-MaP-TAZ and used as the acceptor-only sample. Then the FRET images of the full field of view (FOV) were processed using the PixFRET plugin of ImageJ54. The net FRET is calculated according to equation (5):
$$\mathrm = \frac} - I_} \times \mathrm_} - I_} \times \mathrm_}}}} \times I_}} }}$$
(5)
Where IFRET, Idonor, and Iacceptor are intensities in the region of interest (ROI) under FRET, sfGFP, and MaP microscopy settings, respectively. BTdonor is a factor of the percentage of sfGFP bleedthrough, and BTacceptor is a factor of the percentage of MaP bleedthrough under the FRET microscopy settings. The values for the bleedthrough were determined by analyzing images of donor-only or acceptor-only samples and quantifying the relative intensity ratio under the FRET/donor or FRET/acceptor settings. The ratio was reported as normalized FRET ratio by comparing the net FRET ratio of Halo-MaP-TAZ-labeled cells and Halo-MaP-Me-labeled cells, which do not show ratiometric response to CoA concentrations, according to equation (6).
$$}\,} = \frac_}}}_}}} \times 100}}}$$
(6)
The ROI was defined for each FOV by thresholding the fluorescence intensities at the GFP channel to identify more than 50 cells per FOV. The normalized FRET values for the ROIs (cells) were extracted and used for further calculations and statistical analysis.
Intracellular labeling efficiencyThe complete labeling of intracellular protein was determined by in-gel fluorescence. HEK293 cells expressing the cytosolic Halo-SNAP fusion protein were labeled with 1 μM Halo-MaP-TAZ in full growth medium for 2, 4, 6, 8, 10, and 12 h. Cells were washed three times with PBS to remove the excess of dyes and were resuspended in PBS supplemented with protease inhibitor cocktail (complete-EDTA-free, Roche). Cells were lysed by two flash freeze–thaw cycles and 1 μM Halo-Alexa488 (Promega) and 1 μM BG-SiR55 was added to quantify the unlabeled fraction of HaloTag. After 30 min incubation, the cell extract was centrifuged for 10 min at 20,000g and the clarified lysate was collected in new tubes and kept on ice. The controls for quantitative labeling were prepared by dually labeling the recombinant sensor with Halo-MaP-TAZ/BG-SiR or Halo-Alexa488/BG-SiR. The different samples were resolved by SDS-PAGE. In-gel fluorescence of Alexa488 (Cy2 channel), MaP (Cy3 channel) and SiR (Cy5 channel) was measured on an Amersham Typhoon 5 Biomolecular Imager (GE Healthcare Bio-Sciences Corp). For quantification, the integrated values of background-corrected band intensities were measured with ImageJ. Labeling efficiency (LE) determination was performed according to equation (7):
$$\mathrm\left( }}} \right) = 100 \times \left( }/S_}}}}/C_}}}} \right)$$
(7)
Where S corresponds to the sample fluorescence intensity of intracellularly labeled protein at the defined collection window (Cy2 and Cy5 channels) and C corresponds to the control fluorescence intensity of purified and labeled protein at the defined collection window.
Complete labeling of intracellular sensor protein was determined by ratiometric imaging in living cells. HEK293 cells expressing the subcellular sensor proteins were labeled with 1 μM Halo-MaP-TAZ in full growth medium for 2, 4, 6, 8, 10, and 12 h. At each time point, the cells were washed with HBSS (0.2 mg ml−1 BSA) once and incubated in this buffer for imaging. The results were presented in Extended Data Fig. 7 and Supplementary Figs 3 and 4.
Protein overexpression in HEK293 cellsCells were transfected with plasmids using Lipofectamine 3000 (Thermo Fisher Scientific). After 12 h, the medium was changed to fresh one and the expression of sensor protein was induced by treatment with doxycycline at 37 °C, 5% CO2 for 12 h. Subsequently, the cells were labeled for another 12 h. Cells transfected with empty vector were used as a negative control.
Knockdown of gene by esiRNAHEK293 cells were reversely transfected with endoribonuclease-prepared short interfering RNA, (esiRNA, Sigma-Aldrich) for the gene of interest or non-targeting esiRNA for firefly luciferase (FLUC) using Lipofectamine RNAiMAX (Thermo Fisher Scientific). 0.3 μl of esiRNA and 0.3 μl of Lipofectamine RNAiMAX reagent were mixed in 10 μl Opti-MEM medium, incubated for 10 min and transferred into 96-well plate. One hundred microliters of the prepared cells (3 × 105 cells per milliliter) was added. After 42 h incubation, the medium was changed to fresh one and the sensor expression was induced by treatment with doxycycline at 37 °C, 5% CO2 for 12 h. Subsequently, the cells were labeled for another 12 h. The cells transfected with non-targeting siFLUC were used as a negative control.
RNA extraction and real-time PCRTotal RNA was isolated from the cells (80–90% confluence) with the RNeasy Mini Kit (QIAGEN). RNA quantity was measured with the Nanodrop (Nanodrop Technologies). RNA was directly used in real-time PCR with the one step SYBR Green Quantitative RT-PCR Kit (Sigma-Aldrich) system. Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control. All the real-time values were compared using the CT method, where the amount of cDNA (gene overexpression or knockdown) was normalized to the housekeeping gene GAPDH (ΔCT) before being compared with the amount of cDNA without treatment (ΔΔCT), which was set as the calibrator at 1.0 (ref. 56). The data represents an average of three independent replicates.
Treatments of HEK293 cells with HoPan, PPanSH, and PZ-2891HEK293 cells stably expressing sensor proteins were transfected with the pcDNA5/FRT plasmid encoding PANK3 gene using Lipofectamine 3000. After 12 h, the cells were incubated in customized vitamin B5-free DMEM (Cell Culture Technologies) supplemented with 1/100 dilution of GlutaMAX-I (Gibco), 1 mM pyruvate (Gibco), 10% (v/v) heat-inactivated FBS (Gibco) with doxycycline for induction for 12 h. At the same time, HoPan (Toronto Research Chemicals) was added to the medium to a final concentration of 400 μM, in either presence or absence of 100 μM PPanSH (Chiralix) for 12 h. Subsequently, the cells were labeled for another 12 h with the indicated compounds.
For the PZ-2891 (Selleckchem) treatment, HEK293 cells stably expressing sensor proteins were treated with 1.0 μM PZ-2891 (2 mM stock in DMSO, 2,000×) in normal growth medium for 12 h with doxycycline for induction. Subsequently, the cells were labeled for another 12 h in the presence of 1.0 μM PZ-2891.
Flow cytometry measurementsTo measure intracellular [ATP] changes, the ATeam sensors were used57. ATeam sensors were transiently expressed in HEK293 cells either localized to the cytosol or the inner membrane of mitochondria. The treatment with 1.0 μM PZ-2891 was performed for 12 h during the transfection of the sensors. Subsequently the medium was exchanged and the cells were treated with 1.0 μM PZ-2891 for additional 12 h. Then the cells were washed and resuspended in PBS containing 2% FBS (FACS buffer). The 10 mM 2-DG treatment was performed 24 h after transfection whereby the cells were washed with growth medium without glucose for 30 min before the treatment. The cells were resuspended in 10 mM 2-DG prepared in FACS buffer and incubated for 30 min before analysis. The experiments were measured at the BD LSRFortessa X-20 Flow Cytometer (Becton, Dickinson and Company) using the software BD FACSDiva. For each replicate 8,000 events were analyzed. The following settings were used to record the donor, FRET and acceptor fluorescence: BV421 (excitation 405 nm; emission 450/50 nm) for CFP channel, BV510 (excitation 405nm; emission 525/50) for FRET channel and FITC (excitation 488 nm; emission 530/30 nm) for YFP. Gating strategy involved the removal of dead cells and debris (SSC-A versus FCS-A) and selection of the cell population expressing the sensors (CFP versus YFP). The gated populations in the different conditions were analyzed by determining the mean of their FRET/CFP ratio. The final results are presented as violin plots from two independent biological experiments (Supplementary Fig. 12).
To measure intracellular [CoA] changes, the cytosolic CoA-SnifitV97T and mitochondrial CoA-SnifitG41S were used. HEK293 cells stably expressing sensor protein were plated in 12-well plates and cultured in full growth medium at 37 °C, 5% CO2. For PanK3-overexpression, the cells were transfected with a plasmid encoding PANK3 gene and the empty plasmid was used as a negative control. For the PZ-2891, HoPan, or PPanSH treatments, the cells were directly incubated with the 1.0 μM PZ-2891, 400 μM HoPan, or 100 μM PPanSH, respectively. Then, the cells were washed once with FACS buffer and were resuspended in this buffer. Ten thousand cells were analyzed on a FACSMelody Cell Sorter (BD Biosciences). The following settings were used to record the donor, FRET and acceptor fluorescence: FITC (excitation 488 nm; emission 527/32 nm) for GFP channel, PerCP (excitation 488 nm; emission 700/54 nm) for FRET channel and PE-Cy5 (excitation 561 nm; emission 697/58 nm) for MaP channel. HEK293 cells were used as blank control. The data was analyzed in FlowJo software. Gating strategy involved the removal of dead cells and debris (SSC-A versus FCS-A) and selection of the labeled cell population (GFP versus MaP). The gated populations in the different conditions were analyzed by determining the mean of their GFP/MaP ratio. The final results are presented as violin plots from three independent biological experiments (Supplementary Fig. 14).
Cell permeabilization testTo quantify the free CoA concentration in cells, we attempted to use fluorescence ratiometry as described by Cambronne et. al.58. However, we found that treating the cells with low concentrations of digitonin led to low efficiency of permeabilization for the cells, whereas higher concentrations led to obvious cell death and leakage of the sensors out of the cells. We then turned to another permeabilizing agent, hemolysin, which can permeabilize the cells with 1–2 nm pores to permit rapid flux of ions and nucleotides, but not of proteins59. We then treated the cells stably expressing the cytosolic sensors with 1 μg ml−1 hemolysin for 40 min, and then equilibrated the cells with certain concentrations of CoA in DPBS buffer. The results showed that the normalized ratio was not stable over 30 min (Extended Data Fig. 10a–c), indicating that it is difficult to maintain the free cytosolic CoA concentration at a defined value.
Live-cell quantification of CoA by FLIMHEK293 cells stably expressing sensor protein, HeLa, U-2 OS, and HepG2 cells were plated in tissue-culture-treated 96-well imaging plates and cultured in full growth medium at 37 °C, 5% CO2. HEK293 cells were induced with doxycycline for 12 h. HeLa, U-2 OS, and HepG2 cells were transiently transfected with the pcDNA5/FRT plasmids encoding the cytosolic apo-CoA-SnifitG41 or mitochondrial apo-CoA-SnifitG41S for 24 h. The sensor constructs were labeled with Halo-MaP-TAZ for 12 h in full growth medium. The cells were washed once with HBSS (0.2 mg ml−1 BSA) before imaging. Fluorescence lifetime measurements were performed on Leica TCS SP8 confocal microscope equipped with a 40× water-immersion objective and a TCSPC module. As excitation source, the white light laser was set to 480 nm with 40 MHz pulse frequency collecting 1,000 photons per pixel. The FRET donor emission was measured on a hybrid photodetector for single-molecule detection with a detection window of 490–540 nm. The images were typically acquired with 512 × 512 pixels, pixel size 0.379 μm, scan speed 400 Hz. All measurements were performed at 37 °C, 5% CO2. Data acquisition and analysis were performed using Leica Application Suite X (LAS X). The fluorescence decays of individual cells were extracted by ROIs (sum of the photons of all the pixels of a ROI, typically with 106 photon counts, total cells > 50) and were fitted using a triexponential decay models (n-exponential reconvolution, χ2 < 1.2).
To set up the calibration curves for CoA concentration, CoA-SnifitV97T, CoA-SnifitG41, and CoA-SnifitG41S were diluted in PBS (25 mM HEPES, 1 mM Mg2+, 1 mM ATP, pH 7.4 for CoA-SnifitV97T and CoA-SnifitG41; pH 8.0 for CoA-SnifitG41S) with a concentration of 200 nM. The fluorescence lifetime was measured in the presence of increasing concentration of CoA and analyzed using LAS X FLIM/FCS (v.3.5.6). An example of fluorescence decays and fitting results were presented in Extended Data Fig. 10e. The amplitude weighted average lifetimes τ were used to calculate the FRET efficiencies according to Equations (8) and (9).
$$E = 1 - \frac}}}}$$
(9)
$$\left[ } \right] = c_\frac}}}} - E}}$$
(10)
τFRET and τD represent the amplitude weighted average lifetimes for the FRET and donor-only sample (unlabeled sensor protein). The lifetime was measured and reported in Supplementary Table 5, respectively. [CoA] was quantified using equation (10), where E, Emin, and Emax correspond to the FRET efficiency of the sensor in situ prior treatment (basal state), in the absence and presence of CoA. c50 is the CoA concentration corresponding to half of the maximum sensor response determined from in vitro titrations at 37 °C.
Liquid chromatography–tandem mass spectrometry measurements for total CoA in cellsThe cells were incubated and treated according to the protocols as described above. A total of 2–8 × 106 cells were collected by centrifugation. The cell pellets were lysed by thorough mixing with 200 μl 80% ethanol, which was pre-mixed with 2.5 μM internal standard (CoA-MA, Supplementary Scheme 3). The mixture was vortexed for 5 min and subsequently, the cell debris and protein aggregates were separated by centrifugation at 20,000g for 10 min at 4 °C. The supernatant was diluted 50 times in a 10 mM HCOONH4 buffer (pH 6.8) for analysis by mass spectrometry.
The analysis was performed using a QT6500+ mass spectrometer from Sciex hyphenated to a Nexera X2 UHPLC from Shimadzu. The instruments were controlled using the Sciex Analyst 1.7 (HotFix 3) software. Data analysis was performed using Sciex MultiQuant 3.0.2 software.
Initially, the analysis of CoA suffered from the lack of a commercially available stable-isotope-labeled internal standard (IS) and the compound instability. Under acidic conditions a drastic CoA signal loss over time was observed, whereas tandem mass spectrometry experiments indicated the formation of dephospho-CoA as a major degeneration product. The degenerative processes were slowed down, resulting in an approximate signal loss of 20% within 8 h, if the samples were dissolved in neutral or slightly alkaline HCOONH4 buffer (15 mM HCOONH4, pH 7.95), permanently cooled (0–4 °C) and separated using chromatography on the basis of an aqueous phase featuring 15 mM HCOONH4 (pH 7.95). In this context, it is worth mentioning that we also found that CoA is temperature labile, resulting in decreased CoA signals with increasing mass spectrometry source temperature. Owing to the compound instability and possible day-to-day variations in the workup procedure, usage of an internal standard was considered as mandatory. Lacking a stable-isotope-labeled CoA analog, a CoA maleimide derivative, CoA-MA, was prepared. The maleimide was incubated in 100 μM GSH to ensure its redox stability. No signal loss was detected within 4 h (data not shown). Furthermore, the chromatographic behavior of the IS compound was evaluated in comparison to CoA. The maleimide featured a retention time of 2.27 min versus a retention time of 2.26 min of CoA, indicating that the IS might also be able to correct electrospray ionization suppression effects.
Freshly prepared samples (10 μl), as described above, were injected on an Acquity UPLC HSS T3 (Waters, 2.1 × 50 mm, 1.8 μm) column. The column temperature was set to 20 °C. The analytes were eluted using a 0.5 ml min−1 flow of an aqueous 10 mM HCOONH4 solution (pH 7.95) and acetonitrile. After sample injection, a 2-min isocratic flow of 98% of the aqueous phase was applied, followed by a 2–95% organic phase gradient in 3 min. Subsequently, the column was washed using 95% acetonitrile and re-equilibrated. For CoA ionization and fragmentation under the described high-flow conditions the following ion source parameters were chosen: curtain gas 40 p.s.i., collision gas ‘medium’, ionization voltage 5,500 V, temperature 450 °C, heater gas 50 p.s.i. and nebulizer gas 80 p.s.i. Tandem mass spectrometry analysis was performed in the multiple reaction monitoring mode. The explicit multiple reaction monitoring transitions were listed in Supplementary Table 6. Samples and calibrators featuring concentration levels at 1 (LOQ), 2.5, 5, 25, 50, and 100 ng ml−1 were run in duplicates. The 10 ng ml−1 calibrator was run four times for statistical evaluation, resulting in coefficients of variation with values <5%. At least five consecutive calibrators were either fitted linear or quadratic with a weighting of 1/x, resulting in the best possible fit in the concentration range of the respective samples.
StatisticsData for in vitro titrations were from three independent replicates and shown as the mean ± s.d. Cell imaging experiments were performed in four independently treated samples (imaging dishes). The normalized FRET ratio was calculated from n = 4, 5, or 6 FOVs for each sample and was used for statistical analysis, which was performed using Microsoft Excel 2016. P values were calculated using two-tailed Student’s t-test. The precise numbers of FOVs (n) and P values for the figures containing imaging data are listed in the Supplementary Data. For all of the imaging experiments, with the exception of the one comprising PanK2ΔN, the entire procedure was performed in three biological replicates repeated three times. Significance was evaluated for each of these three replicates individually as described above. Only if all biological replicates showed significant differences relative to an appropriate control (P < 0.05), the experimental result was considered as significant. One arbitrarily chosen replicate was used for preparation of the figures in the manuscript. The results of all biological replicates are shown in Supplementary Figs. 15 and 16.
Reporting summaryFurther information on research design is available in the Nature Research Reporting Summary linked to this article.
留言 (0)