
記住我










The pathophysiological hallmark of COPD is expiratory flow limitation with consequent air trapping and hyperinflation. The resultant increased mechanical loading of the respiratory system, leading to increased work of breathing, contributes to inability of the respiratory muscles to meet ventilatory drive requirements in many COPD patients. If the ventilatory system response is consistently unable to meet drive requirements, alveolar hypoventilation develops and the ability to expel CO2 is compromised, leading to hypercapnia. Severe and persistent hypercapnia, termed hypercapnic respiratory failure, is defined as chronically high PaCO2 (>45 mmHg) [5]. Hypercapnia can also occur acutely, as when acute hypercapnic respiratory failure develops during exacerbations of COPD [55]. Prior acute hypercapnic respiratory failure is a predisposing factor to the development of chronic hypercapnia [56]. A recent multicentre prospective trial from Germany found the prevalence of daytime hypercapnia to be 25% in COPD patients with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage 3 or 4 disease [57]. Daytime hypercapnia is also a predictor of poor prognosis. In a prospective cohort from China, hypercapnic patients had shorter median survival than normocapnic COPD patients [58].
Chemical, mechanical and direct ventilatory neural mechanisms of chronic hypercapnic respiratory failureVentilatory drive to the diaphragm [59] and other muscles of inspiration (including the scalene and parasternal intercostal muscles) [60] is markedly increased in patients with severe COPD, secondary to complex chemical, mechanical and neural changes. However, the effect of chronic hypercapnia on this elevated ventilatory drive is less well characterised.
Chemical sources of ventilatory drive in hypercapnic patientsCentral chemoreceptors located in the medulla sense decreased pH within the brain extracellular fluid as well as changes in arterial CO2, altering ventilation to maintain pH within a certain range [61, 62]. In contrast, peripheral chemoreceptors within the carotid body and aorta are predominantly sensitive to hypoxia, a sensitivity that is enhanced during conditions of decreased pH [63]. CO2 behaves as an acid in aqueous solution, such that the central and peripheral chemoreceptors have PaCO2 thresholds of ∼45 mmHg and ∼39 mmHg, respectively, after which ventilation increases linearly with increasing PaCO2 [64].
In patients with chronic hypercapnia due to severe COPD, chemoreceptor sensitivity may be decreased [65] (figure 1c). Chronically hypercapnic patients with obstructive airway disease have lower ventilatory responses (i.e. sensitivity) to increased PaCO2 (with and without hypoxia) than normocapnic patients with obstructive disease or healthy controls [65, 66]. It is important to consider, when reviewing these findings, that hypercapnic COPD patients are also typically those with the most severe mechanical limitations [67]. Thus, it is difficult to distinguish to what extent blunting of ventilatory responses within such individuals results from mechanical limitations versus decreased chemosensitivity [68]. When Burgraff et al. [69] simulated hypercapnia in goats through 30 days of exposure to high CO2 levels, they demonstrated no significant alteration in chemoreflex response.
 FIGURE 1
FIGURE 1 Proposed pathway of physiological contributions to hypercapnic respiratory failure. a) Mechanical limitations increase work of breathing and ventilatory neural drive through an increase in airway obstruction, which can result in air trapping and alveolar hyperinflation, as well as diaphragm weakness caused by displacement and molecular changes within the muscle. b) Gas exchange abnormalities include ventilation/perfusion ratio (V′/Q′) mismatch (either a result of obstructed or collapsed airways or compromised pulmonary capillaries) and overall parenchymal and vascular destruction. The increased ventilatory drive and inability to compensate for drive due to mechanical and nonmechanical deficits described eventually lead to chronic elevations in arterial partial pressure of carbon dioxide (PaCO2) (chronic hypercapnia). Chronic hypercapnia itself can then lead to several deficits, including c) a reduction in chemosensitivity and d) reduced efferent signals to the diaphragm from cortical motor centres. Both deficits can in turn increase PaCO2, creating positive feedback loops, which worsen existing hypercapnia.
Neuroplasticity of regions of the brain responsible for ventilatory sensing and neural drive may play a role in the altered responses seen in chronic hypercapnic patients, although this hypothesis has thus far only been explored in animal studies. Within the retrotrapezoid nucleus (RTN; a region containing many chemosensory cells relevant to respiration), expression of several neuropeptides decreases with short-term hypercapnia, but increases with chronically elevated PaCO2 [70]. One of these is galanin, which inhibits ventilatory signals including the acute chemosensory response to hypercapnia and hypoxia when injected into the Bötzinger and pre-Bötzinger complexes of rats [71]. This offers a potential mechanism to explain findings of blunted acute chemosensitivity in hypercapnic patients, although the limitations of applying animal physiology to human physiological function must be considered [70]. However, the neuropeptide neuromedin B, which is an excitatory neurotransmitter potentially implicated in increased minute ventilation, is also expressed in increasing quantities in rat RTN neurons as hypercapnia progresses, which may explain the sustained increase in ventilation during chronic hypercapnia [70].
Mechanical modulation of ventilatory drive in hypercapnic patientsCOPD results in airways obstruction, ventilation/perfusion (V′/Q′) mismatch, respiratory muscle dysfunction and hyperinflation, the latter of which leads to diaphragmatic flattening, impaired length–tension relationship of the diaphragm for force generation and consequent changes in breathing pattern (i.e. decreased VT) [72–75]. Hyperinflation further contributes to greater intrinsic positive end expiratory pressure that must be overcome to generate inspiration [76]. In concert, the increased work of breathing resulting from these mechanical deficits requires increased ventilatory neural drive from the respiratory centres within the brain to maintain or attempt to maintain appropriate ventilation per metabolic load in face of reduced ventilatory capacity (figures 1a and 2) [77, 78]. Interestingly, hypercapnia itself may contribute to the perpetuation of this cycle, as even 21 days of chronic hypercapnia increases airway smooth muscle contractility and constriction in response to acetylcholine through a caspase-7-mediated mechanism in murine models [78]. This hypercapnia-mediated increase in contractility may potentiate airways resistance and load in hypercapnic COPD patients, which in turn could further increase ventilatory drive through mechanosensory afferent pathways [79]. In addition, cellular and molecular changes within the diaphragm contribute to mechanical limitations in chronic hypercapnic respiratory failure. On the level of individual cells, diaphragmatic muscle fibres from patients with severe COPD generate less force than those within the diaphragms of healthy controls (figure 1a) [80]. However, the proportion of slow-twitch fibres within the diaphragm is increased in COPD patients, indicating a potential increase in fatigue resistance to compensate for higher ventilatory load [81]. It is still unclear whether this compensation is able to prevent or delay the onset of hypercapnic respiratory failure; more investigation is needed in this domain.
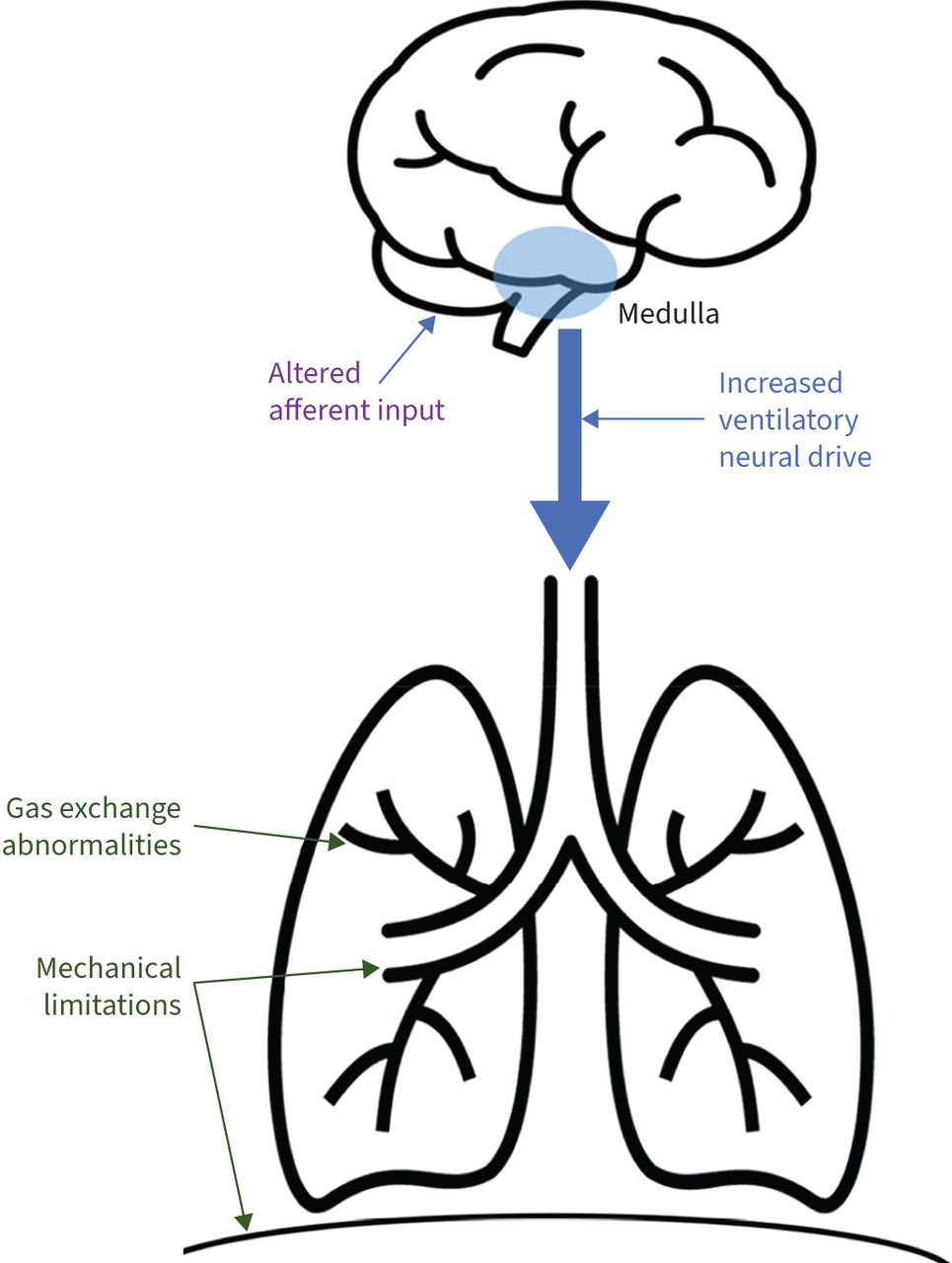 FIGURE 2
FIGURE 2 Physiology of increased ventilatory neural drive in COPD patients with hypercapnic respiratory failure. Gas exchange abnormalities (including ventilation/perfusion mismatch and parenchymal and vascular destruction) as well as mechanical limitations (airways obstruction and respiratory muscle impairment from hyperinflation) ultimately lead to hypercapnia when ventilation is no longer able to match metabolic demand. In turn, altered signalling to the central respiratory centres from chemical and mechanical receptors leads to an increase in ventilatory neural drive to the muscles of respiration.
An additional and significant factor contributing to gas exchange derangement and eventual hypercapnia, which is linked to both mechanical and chemical alterations, is V′/Q′ mismatch (see figure 1b). In early COPD, V′/Q′ inequality presents, in part, through early collapse of small airways and impaired alveolar ventilation prior to larger airways becoming impacted and altering spirometry [73]. Concurrent destruction of pulmonary capillaries increases the proportion of ventilation that enters poorly perfused alveoli (creation of physiological dead space), further contributing to V′/Q′ mismatch. There is also more recent evidence to suggest that some COPD patients may present with a “vascular phenotype” of disease which leads to early V′/Q′ inequality, in which vascular pruning and vascular dysfunction, as opposed to emphysematous destruction of capillary beds, is the predominant contributor to V′/Q′ mismatch as a result of poor perfusion [82]. Finally, the creation of dead space as a result of airway collapse and perfusion limitation leads to a higher required level of minute ventilation in order to facilitate sufficient gas exchange. Patients with severe COPD and mechanical limitation are often unable to meet this requirement and CO2 retention is the result.
Direct neural inputs to the diaphragmDirect neural inputs to the diaphragm via the corticospinal pathway allow for voluntary control of the diaphragm and facilitate nonventilatory activities such as speech [83]. These pathways have been assessed by way of transcranial magnetic stimulation and measurement of resultant motor evoked potentials of the diaphragm [84]. Signalling to the diaphragm initiated in the cortex through the corticospinal pathway contributes to neural drive in the waking state and is essential for resisting apnoeas induced by hypocapnia [85]. This drive is predominantly provided by excitatory stimulation of the cortex via the reticular activating system during wakefulness, allowing for continuation of breathing even without medullary centre input [86].
Evidence suggests that the corticospinal tract is almost maximally activated in awake COPD patients [87] and there is a ceiling effect of the corticospinal signal in COPD patients not seen in healthy controls [88]. However, in COPD patients with elevated PaCO2, findings are inconsistent, with some studies suggesting that corticospinal inhibition of the diaphragmatic motor cortex is increased in hypercapnia [87], while others demonstrating increased voluntary activation of the diaphragm possibly conferring protective advantages in face of worsened mechanics [89]. Such findings have been obtained in relatively small sample sizes, and further work is needed to definitively identify the impact of chronic hypercapnia on ventilatory neural drive. Relative changes in input from medullary respiratory centres may also contribute to changes in drive, but their respective contributions in severe COPD and/or hypercapnia have not been characterised.
Ventilatory neural drive in sleeping patients with hypercapnic COPDDespite persistently elevated daytime ventilatory neural drive in patients with COPD, neural drive changes occurring during sleep may especially predispose patients with impaired respiratory mechanics to nocturnal hypoventilation. To date, EMGdi has been acquired in few overnight studies in patients with COPD. Luo et al. [18] demonstrated greater declines in EMGdi during the transition from wakefulness to NREM and REM sleep in normocapnic COPD versus heathy controls despite consistently higher overall drive in COPD patients, concluding that such decreases in drive may contribute to hypoventilation and hypercapnia during sleep (figure 3a). This is supported by recent findings showing greater loss of EMGdi in the transition from wakefulness to sleep in normocapnic COPD than in health [90]. Such nocturnal hypercapnia is predicted to precede the onset of persistent daytime hypercapnia [91]. As described in the preceding section, corticospinal input to the respiratory centres contributes to neural drive in the waking state, but much of this input is lost during sleep. This has potential to contribute to the drop in EMGdi observed in the transition to sleep. It may also be postulated that the loss of this wakefulness drive could have exaggerated impacts on COPD patients as compared to the healthy population, due to impaired ability to compensate for the increasing mechanical and chemical deficits which we have described in hypercapnic COPD, thus increasing vulnerability to hypoventilation. Interestingly, decreases in EMGdi in the transition to sleep may occur in the presence of preserved ventilatory effort (oesophageal and transdiaphragmatic pressure) in normocapnic COPD [90].
 FIGURE 3
FIGURE 3 Physiological changes during sleep in normal subjects and COPD. a) During sleep, a decrease in ventilatory neural drive is observed. Despite higher baseline drive in COPD patients, a larger drop from wakefulness to sleep is typically noted in this population. b) Increased airway resistance during sleep may exacerbate the ventilation/perfusion (V′/Q′) mismatching experienced in COPD. c) Accessory inspiratory muscle activity is typically eliminated during rapid eye movement (REM) sleep, necessitating a reliance on the diaphragm. However, in COPD patients with compromised diaphragm activity, hypoventilation may result from this loss of accessory muscle activity. d) Finally, decreased chemosensitivity in sleep compounds the existing reduction in chemosensitivity experienced by COPD patients, especially those with elevated arterial partial pressure of carbon dioxide. These changes in sleep compounded on the limitations on COPD patients can contribute to sleep hypoventilation in this population.
In addition to disturbed mechanical inputs to the respiratory centres, disrupted chemical afferent inputs to the central rhythm generator in the medulla in COPD patients during sleep may facilitate hypoventilation and promote CO2 retention (figure 3d). The decreased sensitivity of chemoreceptors to both hypercapnia and hypoxia throughout sleep [92] which causes minimal disturbance in healthy subjects can be deleterious when compounded by the mechanical limitations and blunted chemosensory responses in COPD patients [93]. Furthermore, diaphragmatic flattening due to hyperinflation may cause COPD patients to increase use of accessory inspiratory muscles to maintain ventilation during the daytime and portions of the night [94]. However, REM sleep-associated muscle atonia disproportionately affects inspiratory muscles other than the diaphragm [95, 96], leading to a presumed reliance on the diaphragm to maintain adequate ventilation [97] during REM sleep (figure 3c). This may leave COPD patients who rely in larger part on nondiaphragmatic inspiratory muscles without adequate means to generate pressure during REM sleep, contributing to sleep hypoventilation. Insufficient pressure generation from the diaphragm also encourages patients to adopt a rapid shallow breathing pattern [98], which leads to a higher percentage of ventilation within anatomical dead space, and less efficient gas exchange [99]. Accordingly, hypopnea and associated hypercapnia typically first present during REM sleep [20]. However, emerging evidence suggests that REM sleep atonia of inspiratory muscles apart from the diaphragm may not be as universal as previously believed. In patients with severe COPD recovering from exacerbation, evidence of additional inspiratory muscle activity has been documented during REM sleep [100]. Similarly, maintained activity of other inspiratory muscles, including the parasternal intercostals, has been demonstrated in healthy individuals during REM sleep [101], suggesting that inhibition of inspiratory muscles may be a less significant factor in sleep hypoventilation than once believed.
The prevalence of sleep disordered breathing, in particular, obstructive sleep apnoea (OSA), is high in patients with severe lung disease [102], which can further exacerbate declines in lung function and derangement of blood gases overnight. High apnoea–hypopnea index scores, which indicate the existence of OSA, have been found to be inversely correlated with forced expiratory volume in 1 s (FEV1)/forced vital capacity, indicating that this condition disproportionately affects severe COPD patients [103]. Increased airway inflammation and larger and more frequent oxygen desaturations may contribute to hypoventilation and eventual hypercapnia in OSA patients. Additionally, COPD patients frequently experience arousal from sleep, which can be made even worse by coexistent OSA [104]. These arousals have been associated with increased neural drive, which could complicate our understanding of ventilatory drive during sleep and changes in blood gasses [26, 70]. However, the details of COPD–OSA overlap and arousals are beyond the scope of this article, as we aim to predominantly review the physiological underpinnings of hypercapnic COPD in isolation.
Treatments targeted towards improving ventilation during sleep include nocturnal bronchodilator therapy and oxygen supplementation. While dual long-acting nocturnal bronchodilators decrease airways resistance and sleeping ventilatory effort and ventilatory neural drive in moderate COPD [105, 106], nocturnal long-term oxygen therapy improves oxygenation in patients with persistent hypoxaemia [107] decreasing minute ventilation through reduction of a chemosensory stimulus [108]. These approaches are often insufficient when treating severe COPD with hypercapnia [109]. Recently, nocturnal NIV has gathered significant interest as an effective means of improving blood gas levels in hypercapnic COPD patients and contributing to improved symptom profile [110].
留言 (0)