
記住我






Phosphatidylinositol 3-kinase (PI3K) is an intracellular lipid phosphokinase and the starting point of the PI3K–protein kinase B (AKT)–mammalian target of rapamycin (mTOR) signaling pathway. It is involved in various cellular functions, such as cell growth, proliferation, differentiation, movement, migration, invasion, intracellular transport, and angiogenesis, which are essential for tumorigenesis.[1]
PI3K can be divided into Type I, Type II, and Type III based on differing structures, functions, and substrate specificity.[2] For example, Type I and Type II are involved in cell signal transduction, while Type II and III play an important role in membrane transportation. Type I PI3K maintains the proliferation and survival of human tumor cells. It is composed of a regulatory and a catalytic subunit and can be further divided into IA and IB.[3] The catalytic subunit of class IA PI3K is one of the p110α, p110β, and p110δ encoded by the PIK3CA, PIK3CB, and PIK3CD, respectively. The regulatory subunit is one of the five isoforms of p85 encoded by the PIK3R1 gene, which are p85α, p85β, p55α, p55γ, and p50α. They are regulated by the upstream growth factor receptor tyrosine kinases. Class IB PI3K is composed of the catalytic subunit p110γ encoded by PIK3CG, and one of two related regulatory subunits activated by G protein-coupled receptors, p101 or p87. p110α and p110β are expressed in different types of human cells, while the expression of p110δ and p110γ is limited to immune and hematopoietic cells.[4] After the activation of the PI3K upstream pathway ligand, it directly interacts with the p85 regulatory subunit and inhibits the binding. This leads to the inhibitory effect of p85 on the catalytic subunit p110, and to the activation of PI3K.[5] Activated PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). PIP3 can then act as a second messenger, by recruiting pyruvate dehydrogenase kinase 1 (PDK1) and AKT to the plasma membrane, and phosphorylating serine 473 through the mammalian target of rapamycin complex 2 (mTORC2), which partially activates AKT in the membrane. This will stimulate PDK1 to phosphorylate threonine 308, which will lead to the complete activation of AKT.[6]
AKT is a member of the AGC (protein kinase A [PKA]/protein kinase G [PKG]/protein kinase C [PKC]) protein kinase family, which has serine–threonine protein kinase activity.[7] It is composed of AKT1, AKT2, and AKT3. They are located on chromosomes 14q32, 19q13, and 1q44, respectively. The downstream targets of AKT can be divided into three groups: apoptotic proteins, transcription factors, and protein kinases, which play key roles in various cellular processes, including cell metabolism and apoptosis.[8] It is worth noting that AKT phosphorylates tuberous sclerosis complex 2 (TSC2), and then activates mTORC1 through phosphorylation, which further promotes tumorigenesis, cell cycle regulation, and apoptosis inhibition.[9]
mTOR is an evolutionarily conserved serine/threonine kinase that is divided into two types[10]: mTORC1, which interacts with raptor protein, and mTORC2, which interacts with rictor protein. mTOR stimulates cell growth and proliferation by promoting various anabolic processes and limiting catabolic processes. mTORC2 mainly controls the actin cytoskeleton and contributes to the complete activation of AKT.[11] mTORC1 can regulate translation. Two related downstream effectors are eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4EBP1) and p70 ribosomal S6 kinase 1 (p70S6K1),[12] which can increase translation activity and participate in cell proliferation.
Tumor suppressor phosphatase and tensin homologs (PTEN) are key negative regulators of the PI3K pathway.[13] They regulate downstream signaling pathways by dephosphorylation of PIP3 to PIP2 and prevent further signal transduction by acting as the main regulator agent of the PI3K pathway. Other PI3K negative regulators also include inositol polyphosphate 4-phosphatase type II (INPP4B)[14] and protein tyrosine phosphatase non-receptor 12 (PTPN12).[15]
PI3K Pathway in NSCLCLung cancer is one of the most common forms of cancer. It is a solid tumor with extremely high incidence and mortality rates, with a 5-year survival rate of approximately 5%.[16] Lung cancer can be divided into two major categories, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for 75–85% of lung cancers. Currently, patients with NSCLC can be treated with surgery, radiotherapy, chemotherapy, immunotherapy, and targeted therapy; however, the prognosis and survival rate remain poor.[17] Only some patients can receive surgical resection, which carries a risk of recurrence after surgery. Radiotherapy can cause DNA double-strand break (DSB) in cancer cells to inhibit their growth, but there are still disadvantages, such as radioresistance, metastasis, and local disease progression. Platinum-based chemotherapy used to be the standard treatment for NSCLC, with a poor response rate (17–32%) and overall survival (OS) (7.4–11.3 months).[18,19] Immunotherapy prolongs OS in patients with negative driver genes, but the effect of immunotherapy in patients with positive driver genes is limited. In 2016, the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved pembrolizumab as the first-line treatment for advanced NSCLC (tumor proprotion score [TPS] ≥50%) with negative epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) mutations.[20]
The emergence of new therapies that target specific genetic changes has altered the prospects for the treatment of NSCLC.[21] The genes in key signaling pathways change, which provides advantages for tumor cell survival and proliferation. EGFR is one of the most altered genes involved in NSCLC, and EGFR tyrosine kinase inhibitors (TKIs) improve response rates, postpone time to progression, and prolong OS of NSCLC patients with EGFR mutations.[22] These therapies have produced significant clinical effects when applied to patients with positive mutations in the target gene. Although the response rate of these drugs in mutation-positive patients has been greatly improved compared with chemotherapy, the tumor eventually progresses due to drug resistance. Recently, new oncogene changes have been discovered in NSCLC, including HER2 exon 20 insertion mutations,[23]RET,[24]ROS1 rearrangements,[25] and genetic changes in the PI3K pathway.
There are multiple mutations in the PI3K pathway in NSCLCThe PI3K pathway is involved in the regulation of various cellular functions. Dysregulation of the PI3K signaling pathway can induce various human diseases, especially cancer. Mutations in the PI3K–AKT–mTOR pathway can be observed in many different cancers, such as glioma, liver cancer, breast cancer, colon cancer, ovarian cancer, stomach cancer, and lung cancer.[26]
In NSCLC, changes in the PI3K pathway often appear in high-grade tumors and advanced diseases, which may be related to the degree of lung cancer malignancy.[27] The dysregulation of this pathway occurs through various mechanisms, including the expansion of different subtypes, changes in important targets in the pathway, loss of PTEN, and changes in cell biological behavior.
Mutations and amplifications of PIK3CA are often found in patients with NSCLC.[28]PI3KCA is located at 3q26 on chromosome 3 and encodes the catalytic subunit of PI3K (p110α). The mutations of PIK3CA mainly exist in exons 9 and 20. Exon 9 of PIK3CA encodes the helical domain of p110, and mutations of E542K, E545K, and E545Q in exon 9 may inhibit the inhibitory effect of the N-terminal Src homology 2 (SH2) domain of p85 on the catalytic subunits of p110, leading to excessive PI3K pathway activation.[29,30] Exon 20 encodes the kinase domain of p110, so a mutation in H1047 of exon 20 may promote constitutive activation of the PI3K signaling pathway. In addition to mutations, the more common PI3KCA change is amplification.[31] The PI3KCA gene is amplified to varying degrees in about 35% of lung squamous cell carcinoma (LUSQ) and about 7% of lung adenocarcinoma (LUAD). As the result of a study of 86 NSCLC cell lines, and 356 resected NSCLC tumor tissues, PIK3CA mutation and expansion were found in 12.8% of NSCLC cell lines and 19.1% of tumor tissues.[32] The results of another study showed that PIK3CA mutations were found in 3.7% of tumor tissues of 1144 patients with NSCLC using next-generation sequencing.[28] Among these mutation-positive patients, E545K mutations of exon 9 (57.1%) were the most common. It is worth noting that the PIK3CA mutation in NSCLC may occur simultaneously with EGFR, KRAS, and ALK mutations.
Genetic changes in the AKT family have also been found in NSCLC. E17K is an activating mutation in the lipid-binding pleckstrin homology (PH) domain of AKT1.[33] It is rare and exists in 1–2% of LUSQ cases. However, studies have shown that there is an overexpression of AKT1 and AKT2 in 19% and 32% of LUSQ cases, and 16% and 12% of LUAD cases, respectively.[27] A study of 110 NSCLC tumor specimens showed that 51% of the tumor tissues had increased AKT activity.[34] It has been reported that there may be a certain correlation between PI3K–AKT dysregulation and the grade or stage of the tumor.[35]
The upregulation of the mTOR pathway has also been confirmed in a large population of patients with NSCLC,[33,36,37] and active phosphorylated mTOR (p-mTOR) is present in 90% of patients with LUAD, 60% of patients with large cell carcinoma, and 40% of patients with LUSQ. The presence of mTOR activity may also be a poor prognostic factor for early NSCLC. Several studies have shown that increased mTOR expression is associated with poor survival.[38]
PTEN is another common gene change in the PI3K pathway in NSCLC, and it is one of the key mechanisms that enhance the PI3K pathway signal to initiate and enhance cancer. The loss of PTEN function may be caused by mutations, deletions, or inhibition of transcription by promoter hypermethylation.[13] According to reports,[26,39] PTEN deletions are present in 8–59% of LUSQ cases, and 4–46% of LUAD cases, while PTEN mutations are distributed in 3–10% of LUSQ cases, and 2–5% of LUAD cases. A series of early NSCLC specimens showed that PTEN expression was completely lost in 44% of tumors, 29% had a reduced expression level, and 27% had a normal expression level. A retrospective analysis of the Phase III FLEX study[40] of chemotherapy combined with cetuximab in patients with EGFR-expressing advanced NSCLC showed that 35% of patients had negative PTEN expression, suggesting that the presence of PTEN expression may be associated with improved survival.
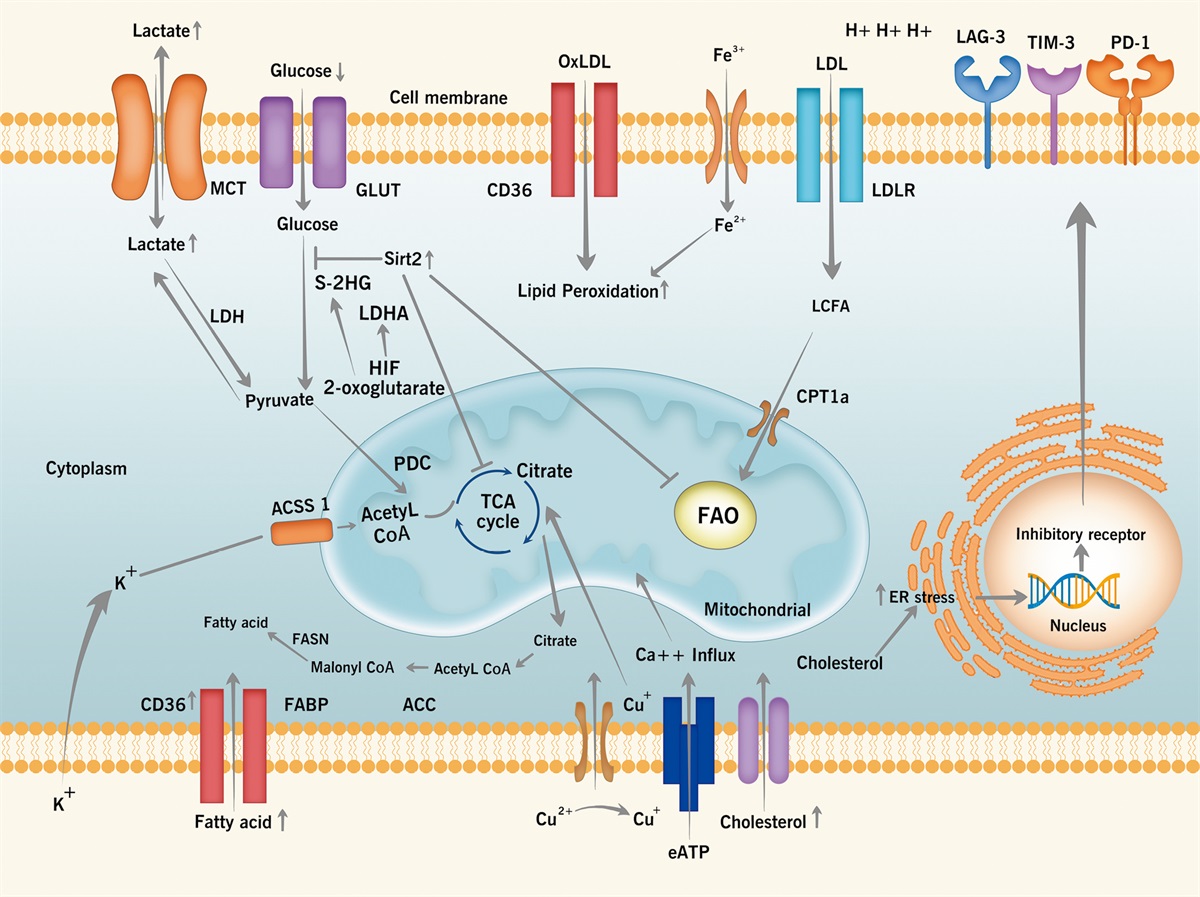
Activating the PI3K pathway promotes the proliferation and metastasis of NSCLCThe PI3K–AKT–mTOR pathway is involved in various cellular processes that promote tumor growth and metastasis, such as cell survival, cell proliferation, autophagy, cell migration and movement, cell metabolism, genome stability, and angiogenesis [Figure 1].
 Figure 1:
Figure 1: PI3K pathway and its role in the occurrence and development of NSCLC. AKT: Protein kinase B; BAD: Bcl-xL/Bcl-2 associated death promoter; CASP: Caspase; CHEK1: Checkpoint kinase 1; FOXO: Forkhead box O; GPCR: G protein-coupled receptor; GSK3: Glycogen synthesis kinase 3; HIF-1: Hypoxia-inducible factor 1; IκB: Inhibitor of nuclear factor kappa-B; IKK: Inhibitor of nuclear factor kappa-B kinase; MDM2: Murine double minute 2; mTORC: Mammalian target of rapamycin complex; NF-κB: Nuclear factor kappa-light chain enhancer of activated B cells; NO: Nitric oxide; NSCLC: Non-small cell lung cancer; P53: Protein 53; PDK1: Pyruvate dehydrogenase kinase 1; PI3K: Phosphosphatidylinositol-3-kinase; PIP2: Phosphatidylinositol 4,5-bisphosphate; PIP3: Phosphatidylinositol-3,4,5-triphosphate; PTEN: Phosphatase and tensin homolog; RHO: Ras homolog; RTKs: Receptor tyrosine kinases; TSC: Tuberous sclerosis complex; VEGF: Vascular endothelial growth factor.
In the PI3K pathway, PTEN is a key regulator for regulating cell cycle progression. Blocking the PI3K pathway can reduce the expression level of S-phase kinase-associated protein 2 (SKP-2) protein in lung cancer cells and block the cell cycle in the G1/S phase.[41] Constitutive activation of AKT or loss of PTEN is one of the most common causes of altered cell survival. The activation of AKT can downregulate the pro-apoptotic B-cell lymphoma 2 (BCL2) family members, BCL2-associated agonist of cell death (BAD) and BCL2-associated X protein (BAX), and promote the activation of apoptosis-related caspase hydrolases, caspase-3 and caspase-9, thereby inhibiting cell apoptosis.[7,42] AKT can also phosphorylate the proto-oncogene murine double minute 2 (MDM2), leading to downregulation of p53-mediated apoptosis.[43] In addition, activated AKT can also block the inhibition of the nuclear factor kappa-light chain enhancer of B cells (NFκB), by members of the inhibitor of nuclear factor kappa-B (IκB) family.[44] NFκB has a wide range of actions and can regulate the expression of hundreds of genes involved in cell apoptosis, cell cycle, cell adhesion, differentiation, and immune regulation.
The PI3K–mTOR pathway negatively controls autophagy through various mechanisms.[45] First, when mTORC1 is inhibited, adenosine 5-monophosphate-activated protein kinase (AMPK) phosphorylates and activates autophagy-promoting UNC-51-like kinase 1 (ULK1), which promotes autophagosome formation. mTORC1 phosphorylates ULK1 at multiple sites or phosphorylates and inhibits ULK1's positive regulators, autophagy-related (ATG)14 and ATG13, to inhibit its interaction with AMPK, thereby preventing AMPK-dependent activation of phosphorylation and preventing autophagy. In addition, transcription factor EB (TFEB) is responsible for genes involved in lysosomal biogenesis. mTORC1 can also indirectly inhibit autophagy through phosphorylation and inhibition of nuclear translocation of TFEB.
The PI3K pathway also plays an important role in cell migration and movement. PI3K and AKT can regulate the epidermal–mesenchymal transition (EMT) of NSCLC.[46] EMT promotes tumor invasion and metastasis and increases cell motility and invasiveness. The loss of PTEN function can change the tumor microenvironment, inhibit the remodeling of the extracellular matrix, and promote tumor cell invasion and metastasis. Ras homolog (Rho) family proteins are involved in the assembly and formation of actin,[47] and are related to membrane folds, cell movement, and cell proliferation. mTORC1 can regulate the activity of Rho family GTPases, thereby changing cell migration and invasion capabilities. Additionally, mTORC1 has been shown to upregulate matrix metalloproteinase 9 (MMP-9) for the proteolytic digestion of extracellular matrix,[48] thereby regulating F-actin reorganization, focal adhesion formation, and tissue remodeling, and further regulating cell migration and invasion.
More importantly, the activation of the PI3K–AKT pathway regulates tumor angiogenesis through multiple downstream effectors, such as mTOR, forkhead box O3 (FOXO3), nitric oxide synthase (NOS), and glycogen synthesis kinase 3 (GSK3).[49,50] These effectors usually upregulate the expression of hypoxia-inducible factor 1 (HIF-1), thereby stimulating transcriptional activation of vascular endothelial growth factor (VEGF), a powerful stimulator of neovascularization, and promote tumor angiogenesis. Additionally, the PI3K–AKT signaling pathway can also upregulate the expression of tumor necrosis factor (TNF),[51] promote endothelial cell migration, and regulate tumor angiogenesis.
The Warburg effect refers to the conversion of glucose into lactic acid by cancer cells through high-rate anaerobic glycolysis,[52] which produces a large amount of energy and biological macromolecules. The PI3K pathway plays an important role in this process. On one hand, inhibiting the PI3K pathway prevents the activation of Tre-2/BUB2/cdc 1 domain 4 (TBC1D4) to increase the translocation of the glucose transporter 4 (GLUT4),[53] and prevents gluconeogenesis by inhibiting FOXO1 and peroxisome proliferator activated receptor gamma coactivator-1 alpha (PPARGC1A). On the other hand, inhibiting the PI3K pathway can also inhibit cholesterol biosynthesis and GSK3, which are necessary for fatty acid uptake and biosynthesis,[54] stimulate the activation of sterol regulatory element-binding protein 1C (SREBP1C), and inhibit the synthesis of cholesterol and fatty acids. Additionally, the lack of PTEN has been shown to promote adipogenesis and β-oxidation by increasing the levels of peroxisome proliferator activated receptor gamma (PPAR-γ) and SREBP1,[55] and by accelerating the production of energy and biological macromolecules.
PTEN plays a powerful role in maintaining genome stability and DNA repair in the nucleus.[56] Checkpoint kinase 1 (CHEK1) is a protein kinase that regulates DNA damage response and cell cycle checkpoint response. Deletion of PTEN will inhibit CHEK1 and cause genome instability. In addition, nuclear PTEN upregulates RAD51 recombinase,[57] which is a component of the DNA DSB repair system. Therefore, PTEN loss can cause homologous recombination defects in human tumor cells and damage to the DNA repair system.
PI3K signaling pathway affects the sensitivity of NSCLC to chemotherapy drugs and EGFR–TKIsThe dysregulation of the PI3K signaling pathway is closely related to resistance to radiotherapy, chemotherapy, and hormone-targeted therapy. Cisplatin and its analogs are one of the standard treatment options for the initial treatment of NSCLC. However, many patients develop resistance to chemotherapy in a short period of time. Chemotherapy resistance is a multifactorial phenomenon, in which the dysregulation of the pro-apoptotic and anti-apoptotic pathways plays an important role.[58] In lung cancer, cells become resistant to cisplatin by AKT gene amplification and overexpression. Several studies have confirmed that the PI3K–AKT signaling pathway can regulate anti-apoptosis-related proteins and is closely related to NSCLC chemotherapeutic drug resistance.[59,60] In addition, studies have shown that activation of p53 reverses the resistance of NSCLC cells resistant to chemotherapy with cisplatin by downregulating the PI3K signaling pathway and promoting the production of intracellular reactive oxygen species (ROS).[61]
In addition to chemotherapy, targeted therapy is one of the most important methods for the treatment of NSCLC. There is constitutive activation of the PI3K pathway in 67% of NSCLC patients with EGFR mutations.[62] Clinical studies have shown that overexpression of the PI3K pathway in advanced NSCLC is a poor prognostic factor. Patients with EGFR mutations with PI3K pathway activation had shorter progression-free survival (PFS) and OS.[63] In patients with NSCLC treated with EGFR–TKI, mutations in the PIK3CA gene may be an important indicator for predicting and evaluating patient response and prognosis.[64] Various resistance mechanisms of EGFR–TKI treatment have been reported, among which PI3K pathway changes are one of the most studied.[65] In NSCLC, the absence of PTEN is associated with poor clinical outcomes and resistance to many anticancer drugs including gefitinib and erlotinib.[66,67] Everolimus is an mTOR inhibitor that can overcome resistance to EGFR inhibitors.[65] Part of the resistance mechanism may involve the activation of related pathways caused by negative feedback interruption of the PI3K signaling pathway. AKT can phosphorylate FOXO1, leading to drug resistance.[68] Additionally, AKT can also mediate PIKfyve phosphorylation,[69] promote EGFR transport and degradation, and subsequently reduce EGFR levels, resulting in weakened sensitivity to EGFR inhibitors. The PI3K signaling pathway may have an important regulatory role in NSCLC resistance. Therefore, overcoming PI3K pathway-related drug resistance may increase the clinical benefit of EGFR–TKIs.
PI3K signaling pathway affects NSCLC brain metastasisThe most common distant metastasis site of lung cancer is the brain, with a 20%–40% incidence rate.[70,71] Without treatment, patients with brain metastases (BM) will survive for up to 1–2 months after metastasis. Although treatment for BM of NSCLC has made progress, the efficacy of all treatment methods is low. The process of NSCLC BM is very complex,[71] involving the shedding of lung tumor cells to form circulating tumor cells, which migrate through the blood circulation and pass through the blood–brain barrier (BBB) to establish and grow new tumors in the brain tissue. In recent years, some studies have identified the key molecules involved in this process and their impact on BM. A retrospective study of 61 patients who underwent surgical resection of primary NSCLC and BM showed that changes in genes encoding the PI3K signaling pathway were enriched in BM,[72] indicating that the PI3K pathway may be associated with an increased risk of metastasis. The BM-free survival of patients who have activated PI3K signals in primary NSCLC tumor tissues is significantly shorter, and their disease spreads to the brain significantly faster than patients without PI3K activation. This study highlights the significant correlation between PI3K signaling and the increased risk of metastasis in NSCLC patients.
Research of PI3K Inhibitors in NSCLCThe PI3K–AKT–mTOR signaling pathway has attracted widespread attention as a single or combined target for the treatment of cancer in the past few decades. In 2014, idelalisib (Gilead Sciences) became the first PI3K inhibitor approved for marketing, and it is mainly used for specific B-cell malignancies.[73] Subsequently, in 2017, the pan-I PI3K inhibitor copanlisib (Bayer)[74] and the dual PI3Kδ/PI3Kγ inhibitor duvelisib (Verastem)[75] were approved in 2017. PI3Kα inhibitor alpelisib (Novartis)[76] was approved for the treatment of advanced breast cancer in 2019 and umbralisib (TG Therapeutics)[77] for the treatment of chronic lymphocytic leukemia, follicular lymphoma, and marginal zone lymphoma in 2021 has also been approved for listing. In addition to these PI3K inhibitors approved by the FDA for clinical treatment, there are several new PI3K inhibitors for NSCLC in various experimental stages. Targeting the PI3K pathway has provided promising preclinical results,[78] and its efficacy for NSCLC treatment in clinical trials is currently being actively evaluated [Table 1].
Table 1 - Inhibitors of the PI3K–AKT–mTOR pathway in clinical development in NSCLC (as of November 2021). Drug Target In combination with Tumor type Phase State Ref. BKM 120 Pan Class I PI3K Carboplatin and pemetrexed NSCLC Phase I Completed NCT01723800 None NSCLC Phase II Completed NCT01297491 Gefitinib NSCLC Phase I Unknown NCT01570296 Erlotinib NSCLC Phase II Completed NCT01487265 Gemcitabine and cisplatin Advanced solid tumors Phase I Withdrawn NCT01971489 None NSCLC Phase I Completed NCT02128724 MEK162 Advanced solid tumors Phase Ib Completed NCT01363232 GDC-0941 Pan Class I PI3K Either paclitaxel and carboplatin (with or without bevacizumab) or pemetrexed, cisplatin, and bevacizumab NSCLC Phase I Completed NCT00974584 Carboplatin/paclitaxel or carboplatin/paclitaxel/bevacizumab NSCLC Phase II Completed NCT01493843 Erlotinib Advanced solid tumors Phase I Completed NCT00975182 Gedatolisib PI3K/mTOR Paclitaxel and carboplatin NSCLC Phase Ib–II Terminated NCT02920450 Paclitaxel and carboplatin Advanced solid tumors Phase I Completed NCT02069158 Ipatasertib AKT None NSCLC Phase II Recruiting NCT04467801 Everolimus mTORC1 None NSCLC Phase II Completed NCT00124280 None NSCLC Phase I Completed NCT00401778 Atalanib Advanced solid tumors Phase I Completed NCT00655655 Aspirin PI3Kα Osimertinib NSCLC Unknown Not yet recruiting NCT04184921, NCT03543683, NCT03532698 AZD8186 PI3Kβ/PI3Kδ As monotherapy and in combination with abiraterone acetate or AZD2014 Advanced squamous NSCLC, CRPC, TNBC Phase I Completed NCT01884285 XL765 PI3K/mTOR Erlotinib Solid tumors Phase I Completed NCT00777699, NCT00692640 MSC1936369B Advanced solid tumors Phase I Completed NCT01390818 Alpelisib PI3Kα None NSCLC Phase II Completed NCT02276027 XL147 Pan Class I PI3K XL647 Solid tumors Phase I Withdrawn NCT00704392 Paclitaxel and carboplatin Solid tumors Phase I Completed NCT00756847 Idelalisib PI3Kδ Pembrolizumab NSCLC Phase Ib–II Unknown NCT03257722 Buparlisib Pan Class I PI3K Docetaxel NSCLC Phase I/II Terminated NCT01911325 PX-866 Pan Class I PI4K Docetaxel Solid tumors Phase I/II Completed NCT01204099 MK-2206 AKT None NSCLC, SCLC, and thymic malignancies Phase II Active, not recruiting NCT01306045 BGB-10188 PI3Kδ Tislelizumab Solid tumors Phase I/II Recruiting NCT04282018 BEZ235 PI3K/mTOR MEK162 Advanced solid tumors Phase Ib Completed NCT01337765 IPI-549 PI3Kγ Nivolumab Advanced solid tumors Phase I Active, not recruiting NCT02637531 CC-223 mTORC1 Erlotinib or azacitidine NSCLC Phase I Completed NCT01545947 None Advanced solid tumors Phase I/II Completed NCT01177397 Temsirolimus mTORC1 None NSCLC Phase II Completed NCT00079235 Sirolimus mTORC1 Auranofin NSCLC and SCLC Phase I/II Recruiting NCT01737502 Pemetrexed NSCLC Phase II Terminated NCT00923273 Gold sodium thiomalate Advanced squamous NSCLC Phase I Withdrawn NCT01383668 Durvalumab NSCLC Phase Ib Recruiting NCT04348292 Panitumumab mTOR None NSCLC Phase I Completed NCT00352950 ABI-009 mTOR Nivolumab Advanced sarcoma and certain cancers Phase I/II Recruiting NCT03190174 AZD2014 mTOR None NSCLC Phase II Recruiting NCT02664935 Sorafenib mTOR None NSCLC Phase II Completed NCT00098254 DS-3078a mTOR None Advanced solid tumors or lymphomas Phase I Completed NCT01588678AKT: Protein kinase B; CRPC: Castrate-resistant prostate cancer; mTOR: Mammalian target of rapamycin; mTORC1: Mammalian target of rapamycin complex 1; NSCLC: Non-small cell lung cancer; PI3K: Phosphosphatidylinositol-3-kinase; SCLC: Small cell lung cancer; TNBC: Triple-negative breast cancer.
Pan class I PI3K inhibitors have a certain inhibitory effect on all subtypes of PI3K, including GDC-0941, BKM120, PX-866, and XL-147. GDC-0941 mainly inhibits p110α and p110δ subtypes, and shows a synergistic effect with mitogen-activated protein kinase kinase (MEK) inhibitors in the treatment of advanced solid tumors.[79] In a phase IB dose escalation trial, patients with advanced NSCLC received GDC-0941 combined with standard first-line chemotherapy, namely carboplatin and paclitaxel, or cisplatin and pemetrexed, and optionally added bevacizumab.[80] Experimental results showed that 43.9% of patients achieved partial response (PR), and 30.9% of patients had stable disease. However, the results of the phase II study showed no significant prolongation of PFS or OS.[81] BKM120 is another oral pan class I PI3K inhibitor. Compared with wild type (WT)-PIK3CA, BKM120 showed better efficacy in the treatment of cancer patients with PIK3CA mutations.[82] In a phase I trial of 43 patients with relapsed and refractory solid tumors using PX-866 combined with docetaxel, one patient with NSCLC and PIK3CA mutation achieved PR.[83] In a phase I trial of XL-147, either as a single agent or in combination with erlotinib, PR was achieved in one patient for each type.[80,84]
In addition to pan class I PI3K inhibitors, various inhibitors targeting PI3K subtypes have also been studied to increase efficacy and reduce toxicity in the treatment of NSCLC. PI3Kα-specific inhibitors have better anticancer activity in tumors with PIK3CA mutations. Among them, the p110α-specific inhibitors, which are entering clinical trials, mainly include alpelisib[85] and serabelisib.[86] Aspirin has also been found to specifically inhibit p110α.[87] It is reported that PI3Kβ plays an important role in PTEN-deficient cancers.[88] Specific p110β inhibitors mainly include GSK2636771,[89] AZD818670,[90] SAR26030171,[91] and taselisib.[92]
AKT's adenosine triphosphate (ATP) competitive inhibitors include, but are not limited to, GSK-690693, perifosine, and MK2206. GSK-690693 is a new type of AKT inhibitor, which can inhibit the proliferation and induce apoptosis of H460 and A549 NSCLC cells.[93] Perifosine, an AKT inhibitor, produced a PR in a patient in a phase I trial with 15 patients with advanced NSCLC.[94] MK2206 is an allosteric small-molecule inhibitor that can inhibit all AKT subtypes. Its phase I clinical trials for NSCLC have shown promising effects in causing tumor shrinkage, and it is well-tolerated as a monotherapy in patients.[95] In addition, the combined treatment of MK2206 with paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors has also been tested in phase I clinical trials, in which one patient with NSCLC achieved PR.[96] In a phase II trial of MK2206 for NSCLC, the combination with erlotinib was evaluated in patients with NSCLC who had previously progressed on erlotinib therapy. Patients were stratified according to EGFR mutation status. The median PFS of patients with EGFR mutations was 4.4 months, and that of EGFR wild-type patients was 4.6 months.[97]
So far, the mTORC1 inhibitors approved by the FDA for clinical trials in patients with NSCLC mainly include temsirolimus and everolimus, both of which are derivatives of rapamycin. Rapamycin was originally isolated from the soil of Rapa Nui Island and has antifungal activity.[98] Various rapamycin analogs have been developed. Rapamycin binds to FK506 binding protein 12 (FKBP12) to destroy raptor interaction with mTOR, leading to the dissociation and inactivation of the mTORC1 complex to prevent cell proliferation. Temsirolimus, as monotherapy, has a clinical benefit rate (confirmed response and confirmed stable disease) of 35% in newly treated patients with NSCLC.[99] Everolimus can selectively inhibit mTORC1 signaling, but in the phase II trial, when combined with erlotinib, it failed to show significant efficacy in patients with advanced NSCLC.[100] In addition to rapamycin, various ATP-competi
留言 (0)