
記住我



Studies have shown a transient decrease in microglial densities in brains of B6;Cx3cr1−/− mice between post-natal days 8 through 28 [12]. Similarly, sc-RNA seq studies that have demonstrated that transcriptomic differences observed in FACS purified, CD11b+CD45+ microglia from 2 month-old B6;Cx3cr1−/− mice are not evident in aged microglia from 12 and 24 month-old Cx3cr1 deficient mice as compared to cells isolated from age-matched B6;Cx3cr1+/+ animals [31]. These studies imply that Cx3cr1 deficiency has transient and subtle effects on the transcriptional landscape of homeostatic microglia. To ascertain that no overt microglial defects persist into adulthood, we examined 6 month- old B6;Cx3cr1+/+ and B6;Cx3cr1−/− mice for microglial abundance and homeostatic activation. qRT-PCR analyses using cortical mRNA revealed similar expression of canonical microglial genes namely Pu.1, Iba1, P2ry12 and Cd11b (Supplementary Fig. 1Ai-Aiv). Furthermore, flow-cytometry analyses revealed similar numbers of CD11b+ cells in the brains of B6 mice with and without Cx3cr1 (Supplementary Fig. 1B). Interestingly, while no differences were observed in the number of cellular processes between the two genotypes, Cx3cr1−/− microglia displayed a modest increase in the number of process junctions as compared to microglia from B6;Cx3cr1+/+ mice (Supplementary Fig. 1C). Lastly, with the exception significantly reduced levels of Cst7, we observed no significant differences in the expression of genes associated with homeostatic or inflammatory microglia in 6 month-old B6;Cx3cr1+/+ and B6;Cx3cr1−/− mice (Supplementary Fig. 4A, B). Taken together with these published studies, our data show similar homeostatic microglial signatures in 6 month-old B6 mice with and without Cx3cr1.
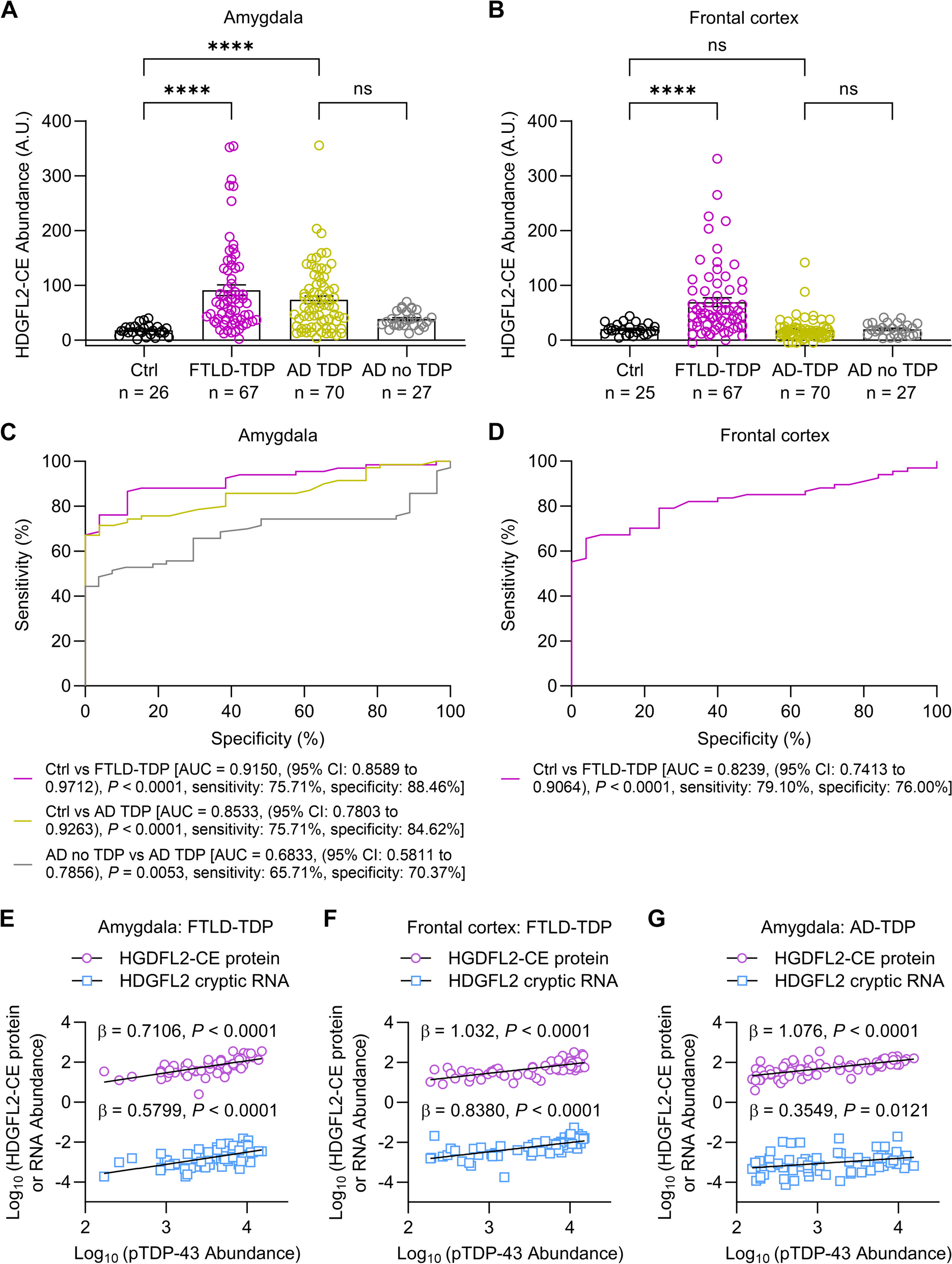
Cx3cr1 deficiency leads to accelerated plaque depositionTo investigate the kinetics of Aβ accumulation in the absence of Cx3cr1, brain sections from 4 and 6 month-old 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice were immunolabeled with anti-Aβ1-42 antibodies. As seen in our previous studies using APPPS1 mice [25, 26], loss of Cx3cr1 resulted in significantly reduced MOAB2+ plaque load in the cortex and hippocampus in 4-month-old 5xFAD mice (Fig. 1A-C). By contrast, at 6 months of age, the number of MOAB2+ plaques were significantly increased in the cortex and hippocampus of 5xFAD;Cx3cr1−/− mice as compared to age-matched 5xFAD;Cx3cr1+/+ animals (Fig. 1A-C). 6-month-old 5xFAD;Cx3cr1+/+ mice showed an ~ 1.5 fold and ~ twofold increase in Aβ42 plaque loads in the cortex and hippocampus respectively as compared to their 4 month-old counterparts. By contrast, an ~ 2.8 fold and ~ sixfold higher plaque burden in the cortex and hippocampus of 6 month of 5xFAD;Cx3cr1−/− mice over 4 month-old 5xFAD;Cx3cr1−/− animals indicated that plaque deposition is accelerated with disease progression in the absence of Cx3cr1 (Fig. 1A-C).
Cx3cr1 deficiency exacerbates the accumulation of neurotoxic species of AβInsoluble aggregates of fibrillar Aβ (fAβ) along with soluble, oligomeric Aβ (oAβ) are associated with neurotoxicity in AD [32,33,34]. To assess whether the loss of Cx3cr1 alters the accumulation these neurotoxic Aβ species, we first stained serial brain sections with Thioflavin S (ThioS) to visualize fAβ plaques. ThioS+ plaques deposited in 5xFAD;Cx3cr1−/− mice appeared significantly more diffuse when compared to those in 5xFAD;Cx3cr1+/+ mice (Fig. 1D). Using circularity analysis, to distinguish compact plaques from plaques with a filamentous/diffuse or an intermediate phenotype (Fig. 1E), we observed that Cx3cr1 deficiency resulted in a significant reduction in the proportion of compact plaques in the cortex (Fig. 1F) and hippocampus (Fig. 1G) of 6-month-old 5xFAD mice, with a concomitant increase in accumulation of plaques with intermediate and diffuse morphologies. To investigate the accumulation of soluble oAβ species, we immune-stained for OC+ oAβ at 4- and 6- months of age. In-situ quantification of OC+ Aβ accumulation revealed that increased proportion of cortical areas were positive of OC immunoreactivity in 5xFAD;Cx3cr1−/− mice throughout the course of the disease (Fig. 1H). Consistent with these results, high-resolution confocal microscopy revealed larger deposits of OC+ oAβ surrounding compact and filamentous ThioS+ plaques in the cortices of 6-month-old 5xFAD;Cx3cr1−/− mice compared to similar plaques in 5xFAD;Cx3cr1+/+ mice (Fig. 1I). While female 5xFAD mice displayed significantly increased plaque burdens compared to males (Fig. 1B, C), no significant differences in OC+ oAβ loads were observed in female and male 5xFAD cohorts. Taken together, our results indicate that Cx3cr1 deficiency shifts Aβ dynamics towards increased accumulation/generation of toxic species of soluble oAβ associated with highly filamentous fAβ plaques.
Microglial engagement of ThioS.+ plaques is compromised in the absence of Cx3cr1Effective microglial proliferation, followed by their recruitment to, and subsequent interaction with Aβ plaques has been associated with trimming of amyloid fibrils and reduced fibril-branching leading to plaque compaction. To investigate whether the shift towards accumulation of diffuse plaques in 5xFAD;Cx3cr1−/− mice correlates with dysregulation of plaque associated microglial responses, we used flow-cytometry and histology to visualize microglia associated with fAβ plaques. Consistent with increased plaque burdens (Fig. 1), we observed increased numbers of CD11b+ microglia in the brains of 6 month-old 5xFAD;Cx3cr1−/− mice (Supplemental Fig. 2Ai-Aiii), which corresponded to a significant increase in the proportion of cortical areas positive for Iba1 (%Iba1+,Supplemental Fig. 2Bi). Furthermore, when %Iba1+ areas were normalized to areas positive for ThioS+ fAβ, we observed no differences in the cortex of 6-month-old 5xFAD mice with or without Cx3cr1 (Supplemental Fig. 2Bii). These data are reflective of efficient microgliosis in response to increased Aβ deposition in 5xFAD;Cx3cr1−/− mice, and similar recruitment of Iba1+ microglia to fAβ plaques in 5xFAD animals regardless of Cx3cr1 genotype. To further investigate Cx3cr1-dependent effects on proliferation of plaque-associated vs. non-plaque-associated microglia, we preformed stereological quantification of Ki67+ microglia in cortical layer V of 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice (Supplemental Fig. 2C-E). 6 month-old 5xFAD;Cx3cr1−/− mice showed a modest but significant increase in the proportion of ThioS+ plaques associated with Ki67+ Pu.1+ Iba1+ microglia (Supplemental Fig. 2D). Additionally, we also observed a significant increase in the proportion of plaque associated Ki67+Iba1+ microglia in 5xFAD;Cx3cr1−/− mice (Supplemental Fig. 2E). No non-plaque-associated Ki67+ Iba1+ cells were observed in 5xFAD mice regardless of their Cx3cr1 genotype.
Interestingly, analysis of microglial plaque-engagement revealed that regardless of the plaque compaction phenotype, Iba1+ microglia formed well-defined barriers at the Aβ interface in 6 month-old 5xFAD;Cx3cr1+/+ mice (Fig. 2A). By contrast, Iba1+ plaque barriers appeared disorganized in 5xFAD;Cx3cr1−/− cohorts (Fig. 2B). Next, we assessed whether Cx3cr1 deficiency altered microglial plaque engagement throughout disease progression. 4 month-old 5xFAD;Cx3cr1−/− mice showed significant reduction in Iba1+ process engagement of filamentous and intermediate ThioS+ plaques when compared to similar plaques in 5xFAD;Cx3cr1+/+ mice. No differences were observed in Iba1+ engagement of compact plaques in the presence or absence of Cx3cr1 at this age (Fig. 2C). Interestingly, at 6 months, while diffuse ThioS+ plaques in 5xFAD;Cx3cr1−/− mice displayed a similar reduction in Iba1+ process engagement, no differences were observed in the proportion of Iba1+ processes associated with intermediate plaques in 5xFAD;Cx3cr1+/+ vs. 5xFAD;Cx3cr1−/− mice (Fig. 2D). By contrast, Cx3cr1 deficiency significantly impaired microglial engagement of compact plaques at this age (Fig. 2D). Studies have shown that TREM2 levels are elevated in the AD brain, and TREM2 expression is enriched particularly in Iba1+ microglial processes that engage with Aβ plaques [28]. Microglial upregulation of TREM2 is not only critical for efficient plaque-engagement, but active TREM2-signaling at the microglia-Aβ interface is also critical for efficient plaque compaction [28]. qRT-PCR analyses of FACS purified, CD11b+ microglia from 6 month-old 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice revealed no differences in expression of microglial Trem2 and its signaling partner, Tyrobp regardless of the Cx3cr1 genotype (Supplemental Fig. 2F). Likewise, we found no differences in the concentration of total TREM2 in the cortical lysates of 6 month old 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice (Supplemental Fig. 2G). Taken together, these results indicate that CX3CR1-signaling shapes microglial plaque interaction without altering microglial proliferation and subsequent recruitment to Aβ deposits.
5xFAD;Cx3cr1.−/− mice show altered apoptotic-signaling, ROS metabolism and oxidative stress responsesGiven the aberrant accumulation of toxic Aβ and impaired microglial plaque engagement in 5xFAD;Cx3cr1−/− mice, we hypothesized that the Aβ driven neuropathological milieu is altered in the absence of Cx3cr1. To investigate how CX3CR1 shapes glial activation and the neuroinflammatory microenvironment, we ran the nCounter® Neuroinflammation Panel which queries 770 genes involved in neuron-glia interactions, inflammation, and neuroplasticity (Supplemental Files 8, 9). Transcriptional analyses using cortical RNA from 6 month-old 5xFAD animals revealed upregulation of genes associated with cellular apoptosis (Casp9, Casp3, Casp8) and pro-survival signaling (Bcl2l1) in 5xFAD;Cx3cr1−/− mice when compared to Cx3cr1+/+ counterparts (Fig. 3Ai-Aii). Interestingly, we observed increased expression of pro-inflammatory genes (Ccl2, Ccl5), along with increased Cst7, P2ry17 and Tgfbr1 levels in 5xFAD;Cx3cr1−/− mice (Fig. 3Ai-Aii). Lastly, genes associated with nitric-oxide signaling, ROS production and oxidative stress responses were differentially altered in 5xFAD;Cx3cr1−/− animals (Pten, Pink1, Nostrin, Sod2, Anxa1, Lcn2) (Fig. 3Ai-Aii). Gene-ontology analysis of differentially expressed genes (DEGs) revealed that in comparison with their female counterparts that displayed regulation of programmed cell death/apoptosis, regulation of ROS metabolism and regulation of TGFβ3 signaling as the key biological pathways affected by the loss of Cx3cr1, top processes affected in male 5xFAD;Cx3cr1−/− mice were associated with signaling related to cell cycle arrest in response to DNA damage and ER stress (Supplemental Fig. 3A). Despite these differences, common pathways altered in female and male 5xFAD;Cx3cr1−/− mice indicated increased cellular apoptosis/necroptosis, altered oxidative/ER stress, increased DNA damage and cell cycle arrest (Supplemental Fig. 3B). Additionally, KEGG enrichment analysis revealed that loss of CX3CR1 signaling may result in altered phagocytosis along with alterations in key signaling pathways with known involvement in intracellular protein shuttling, protein phosphorylation, cellular senescence, synaptic plasticity/transmission, and cognition (Supplemental Fig. 3C).
Cx3cr1 deficiency drives dysregulated microglial activation in 5xFAD miceTo investigate whether the transcriptomic changes identified using the nCounter® system are specifically reflective of microglial dysregulation, we used FACS-purified CD11b+ microglia isolated from brains of 6 month-old 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− animals to validate top DEGs and biological pathways using qRT-PCR. Based on increased P2ry12 expression in male 5xFAD;Cx3cr1−/− mice (Fig. 3Aii), we first investigated the skewing of microglia towards a ‘disease-associated’ phenotype. While Clec7a and Cst7 mRNA levels were upregulated in microglia from male and female 5xFAD;Cx3cr1−/− mice (Fig. 3Bii, Biii), Apoe mRNA levels were elevated in microglia only in female 5xFAD;Cx3cr1−/− mice, when compared to cells isolated from sex-matched 5xFAD;Cx3cr1+/+ mice (Fig. 3Bi). Interestingly, microglia from 5xFAD;Cx3cr1−/− mice showed increased mRNA levels for Tgfβ-r1 with reduced Tgfβ1 expression (Fig. 3Biv, Bv), signaling components that have been implicated in a protective microglial phenotype [35]. Furthermore, microglia from 5xFAD;Cx3cr1−/− brains express significantly increased mRNA levels of pro-inflammatory Ccl2, Ccl5 and Il-1β, along with reduced Tnf and Il-6 transcript levels (Fig. 3Ci – Cv). Taken together, these data indicate that Cx3cr1 deficiency dysregulates microglial activation towards a phenotype primed for increased neurotoxicity. Lastly, increased mRNA levels of Pten (Fig. 3Di) and Cybb/iNos (Fig. 3Dii) coupled with reduction in Pink1 (Fig. 3Diii) mRNA expression indicate that 5xFAD;Cx3cr1−/− microglia display increased oxidative/mitochondrial stress responses. No significant differences in the expression of these genes in 6 month-old B6;Cx3cr1+/+ and B6;Cx3cr1−/− mice (Supplemental Fig. 4) further confirmed that these differences in microglial activation elicited by Cx3cr1 deficiency are indeed driven by AD pathology, and are not observed in the healthy, adult brain. Thus, the loss of CX3CR1-signaling exacerbates microglial dysfunction in AD, driving neurodegenerative activation.
Cx3cr1 deficiency impairs microglial Aβ phagocytosis and lysosomal activityWhile our previous studies have shown that a deletion of Cx3cr1 in B6 mice augments microglial uptake of exogenously injected Aβ42 [25], the role of CX3CR1 in microglial phagocytosis of endogenously produced, extracellular fAβ plaques in the context of accumulating AD pathology is largely unclear. Plaque accumulation in 5xFAD mice in the presence and absence of Cx3cr1 (Fig. 1) suggests that while microglia may be efficient phagocytes in early disease, their Aβ uptake and clearance capacities are impaired with disease progression. Thus, to investigate whether the phagocytic and lysosomal dysfunction suggested by our transcriptomic data (Supplemental Fig. 3) was functionally evident in 5xFAD mice, we injected (i.p.) 4- and 6-month old 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice with methoxy-X04 and analyzed microglial uptake of endogenous fAβ using flow cytometry. Characterization of CD11b+ microglia based on their surface expression of CD45 (Supplemental Fig. 5A), revealed that while CD11b+ CD45low microglia accounted for ~ 35% of fAβ+ phagocytic microglia (Supplemental Fig. 5C), ~ 70% of CD11b+ CD45high microglia were fAβ+, indicating that CD11b+ CD45high cells represent an activated, highly phagocytic microglial subpopulation in 5xFAD mice (Supplemental Fig. 5C). The absence of any fAβ+ microglia in methoxy-X04 treated B6 mice (Supplemental Fig. 5B) indicated that the fAβ+ populations observed in 5xFAD cohorts were indeed microglia that had actively phagocytosed fAβ.
We observed a modest increase in the proportion of fAβ+ CD11b+ microglia in 4 month-old 5xFAD;Cx3cr1−/− mice (~ 44%) as compared to age-matched 5xFAD;Cx3cr1+/+ cohorts (~ 36%), corresponding to a mild, but significant increase in the fAβ+ CD11b+ CD45high microglial subset in these animals (Supplementary Fig. 6A), suggesting increased microglial uptake of fAβ in 4 month-old 5xFAD;Cx3cr1−/− mice. Interestingly, these microglial subsets were further categorized into methoxy-X04low / fAβlow and methoxy-X04high / fAβhigh subpopulations (Supplemental Fig. 5C). To assess whether these varied microglial populations differed in lysosomal activation indicative of their ability of clearing phagocytosed Aβ, we quantified their lysosomal activity using Lysotracker-DeepRed™ (-DR), which specifically labels all acidified, phagocytic compartments. Significantly higher lysotracker-DR mean fluorescence intensities (MFI) in the fAβhigh subpopulations within the CD45low and CD45high microglia, indicated increased endolytic activation in these subsets as compared to fAβlow populations (Supplemental Fig. 6B, C). Interestingly, while lysotracker-DR MFI for the fAβhigh CD11b+ CD45low microglia in 5xFAD;Cx3cr1−/− mice was significantly higher than the corresponding subset from 5xFAD;Cx3cr1+/+ mice (Supplemental Fig. 6B), lysotracker-DR MFIs were significant reduced in all fAβ+ populations within CD11b+ CD45high microglia in 5xFAD;Cx3cr1−/− mice (Supplemental Fig. 6C). These data indicate that despite modest increases in their phagocytic potentials, microglia in 4 month-old 5xFAD;Cx3cr1−/− mice show significant deficits in their overall endolytic activation.
In contrast to 4-month old cohorts, we observed that ~ 26% of CD11b+ microglia from 5xFAD;Cx3cr1−/− mice had internalized fAβ, as compared to ~ 46% fAβ+ microglia in age-matched 5xFAD;Cx3cr1+/+ mice (Fig. 4A). Furthermore, significant reductions in the fAβ+ CD11b+ CD45low and fAβ+ CD11b+ CD45high microglia in 5xFAD;Cx3cr1−/− mice indicated that Cx3cr1 deficiency profoundly impaired microglial Aβ phagocytosis at this age (Fig. 4C). Higher lysotracker-DR MFI of fAβlow and fAβhigh microglia within CD45low and CD45high populations in 5xFAD;Cx3cr1+/+ mice as compared to microglia from B6;Cx3cr1+/+ controls, indicated increased lysosomal activation of phagocytic microglia in 5xFAD mice (Fig. 4B). Interestingly, lysotracker-DR MFIs were significantly reduced in all microglial populations expect for fAβ+ CD11b+ CD45high microglia in 5xFAD;Cx3cr1−/− mice when compared to similar subsets in 5xFAD;Cx3cr1+/+ cohorts (Fig. 4B, D). Taken together, these results demonstrate that a) endolytic dysfunction in microglia in the absence of CX3CR1-signaling begins early in the course of AD and b) CX3CR1-driven effects on lysosomal activation may be crucial in shaping microglial uptake of Aβ as disease progresses.
Cx3cr1 deficiency leads to severe neuritic dystrophyBased on studies that have demonstrated the association of OC+ oAβ with dystrophic neurites [36], we next assessed whether increased oAβ in 5xFAD;Cx3cr1−/− mice (Fig. 1H-I) results in heightened neuritic dystrophy. Using α-LAMP1, α-nT-APP and α-Ubiquitin to visualize neuritic, ThioS+ plaques, we observed that 4 month-old 5xFAD;Cx3cr1−/− showed significantly reduced numbers of dystrophic neurites in their cortices (Fig. 5A, B). However, 5xFAD;Cx3cr1−/− animals displayed larger foci of severe neuritic dystrophy (Fig. 5C), associated with compact (solid arrows) as well as filamentous (dashed arrows) ThioS+ plaques, when compared to similar plaques in 5xFAD;Cx3cr1+/+ mice (Fig. 5C). Indeed, when dystrophic neurites were quantified based on size distribution, we observed that while a majority of LAMP1+ (Fig. 5D), Ubiquitin1+ (Fig. 5E) and nT-APP+ (Fig. 5F) neurites in the cortex of 5xFAD;Cx3cr1+/+ mice were < 500 µm, dystrophic neurites in 4 month-old 5xFAD;Cx3cr1−/− mice were largely > 500 µm in size. This data demonstrates that 4 month-old 5xFAD;Cx3cr1−/− mice display more severe neurodegenerative changes despite reduced plaque loads (Fig. 1) Similar accumulation of larger dystrophic neurites with severe neuritic pathology was also observed in the cortex of 6 month-old 5xFAD;Cx3cr1−/− mice (Supplementary Fig. 7A – E). Lastly, severe neuritic dystrophy in 5xFAD;Cx3cr1−/− mice at this age was strongly correlated with increased accumulation of filamentous fAβ plaques (Supplementary Table 2). These data indicate that the severity of neuritic dystrophy in the absence of Cx3cr1 may be driven by the profile of toxic Aβ species rather than abundance of Aβ plaques.
Cx3cr1 deficiency aggravates tau pathology in 5xFAD miceTo identify mechanisms of overt neuronal and synaptic loss, we investigated the accumulation of pTau in 5xFAD mice. 6 month-old 5xFAD mice showed increased pTau pathology as compared to age-matched B6 controls (Fig. 6A, B). While intraneuronal AT8+ pTau was observed in cortical layer III (Fig. 6B-i, solid arrows, top panel) and the CA2/CA3 region (Fig. 6B-iii,top panel), AT8+ pTau also accumulated in dystrophic neurites in cortical layer V (Fig. 6B-ii, top panel), and in neuritic plaques in the subiculum (Fig. 6B-iv, top panel) in 5xFAD;Cx3cr1+/+ mice. Interestingly, in addition to intraneuronal AT8+ pTau in the CA2/CA3 in 5xFAD;Cx3cr1−/− mice (Fig. 6B-iii,lower panel), mislocalized pTau also accumulated in axonal projections in their cortex (Fig.6A-i, dashed arrows, lower panel). Moreover, while the majority of AT8+ pTau accumulated as dystrophic neurites around senile, neuritic plaques, 5xFAD;Cx3cr1−/−mice displayed heighted pTau+neuritic dystrophy as compared to 5xFAD;Cx3cr1+/+mice in cortical layer V (Fig. 6B-ii, lower panel) and the subiculum (Fig. 6B-iv, lower panel). Indeed, 6 month-old 5xFAD;Cx3cr1−/− mice showed a significant increase in the areas of the cortex, hippocampus and subiculum positive for AT8+pTau as compared to sex-matched 5xFAD;Cx3cr1+/+ animals (Fig. 6C), indicating aggravated deposition of pathological pTau. We observed an ~ 2-fold increase in the level of total, soluble tau in the cortex of 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice when compared to genotype-matched B6 controls (Fig. 6E, F). Furthermore, we observed a significant increase in the levels of soluble, AT8+ pTau in 5xFAD;Cx3cr1+/+ with respect to B6;Cx3cr1+/+ controls, which were further increased in 5xFAD;Cx3cr1−/− mice (Fig. 6E, F). Interestingly, we found a significant correlation between cortical pTau accumulation and levels of OC+ oAβ (Fig. 1H, i) in 5xFAD;Cx3cr1−/− but not 5xFAD;Cx3cr1+/+ mice (Fig. 6D), in line with studies that have implicated these soluble oAβ species in mislocalization and spread of pTau [37]. Lastly, cortical pTau significantly correlated with compact fAβ plaques in 5xFAD;Cx3cr1+/+ mice (Pearson’s r = 0.82, p*<0.05). In contrast, significant correlation was observed between cortical pTau and intermediate fAβ (Pearson’s r – 0.94, p****<0.00001) and diffuse fAβ (Pearson’s r = 0.82, p**<0.003) in 5xFAD;Cx3cr1−/− animals.
Cx3cr1 deficiency aggravates synaptic dysfunction, neuronal loss and cognitive decline in 5XFAD miceTo characterize how increased toxic Aβ levels (Fig. 1), pTau accumulation (Fig. 6) and dysfunctional microglial activation (Fig. 3) affect neurotoxicity, we analyzed the levels of pre- and post-synaptic elements in 6 month-old cohorts. Interestingly, we observed a modest but significant increase in the expression of pre-synaptic proteins SV2a and Synaptophysin, with a significant reduction in Homer in 6 month-old 5xFAD;Cx3cr1+/+ mice when compared to B6;Cx3cr1+/+ controls (Fig. 7A, C). While loss of pre-synaptic Homer was not observed in age-matched 5xFAD;Cx3cr1−/− mice, no significant differences were seen in Sv2a and Synaptophysin in the 5xFAD;Cx3cr1−/− mice compared to 5xFAD;Cx3cr1+/+ cohorts (Fig. 7A,C). In contrast, 5xFAD;Cx3cr1+/+ mice showed an ~ 1.3 fold reduction in post-synaptic proteins, namely PSD95 and NMDAR1 when compared to B6:Cx3cr1+/+ controls. 5xFAD;Cx3cr1−/− mice showed ~ 1.6 fold reduction in PSD95 and NMDAR1 as compared to B6; Cx3cr1−/− mice, and an ~ 1.2–1.3 fold decrease as compared to their 5xFAD; Cx3cr1+/+ counterparts (Fig. 7A, D). Next, quantitation of NeuN+ neurons in the subiculum, the area of the most robust and chronic Aβ pathology in 5xFAD mice, revealed no differences in overall neuronal numbers in 4 month-old 5xFAD mice with and without Cx3cr1 (Fig. 7B). However, we observed a significant reduction in NeuN+ neurons in the subiculum of 5xFAD;Cx3cr1−/− mice at 6 months when compared to age-matched 5xFAD;Cx3cr1+/+ mice (Fig. 7B), which correlated with increased pTau+ pathology in these areas (Pearson’s r = 0.92, p*** < 0.0001, Fig. 6B-iv, C), Taken together, our data indicates that Cx3cr1 deficiency worsens neurodegeneration and synaptic dysfunction in 5xFAD mice. Finally, to assess whether the loss of Cx3cr1 worsens cognitive decline, we measured spatial working memory in 6 month-old cohorts of B6 and 5xFAD mice with and without Cx3cr1. While there were no differences in total arm entries in 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice, we observed a significant reduction in the number of arm entries in B6;Cx3cr1−/− mice as compared to B6;Cx3cr1+/+ controls (Fig. 7E). Significant reduction in spontaneous alternations between arms was observed in 5xFAD;Cx3cr1+/+ and 5xFAD;Cx3cr1−/− mice when compared to genotype matched B6 controls (Fig. 7E). Moreover, significantly decreased spontaneous alternations in 5xFAD;Cx3cr1−/− mice as compared to 5xFAD;Cx3cr1+/+ cohorts demonstrated that Cx3cr1 deficiency aggravates cognitive impairment in AD.
留言 (0)