
記住我


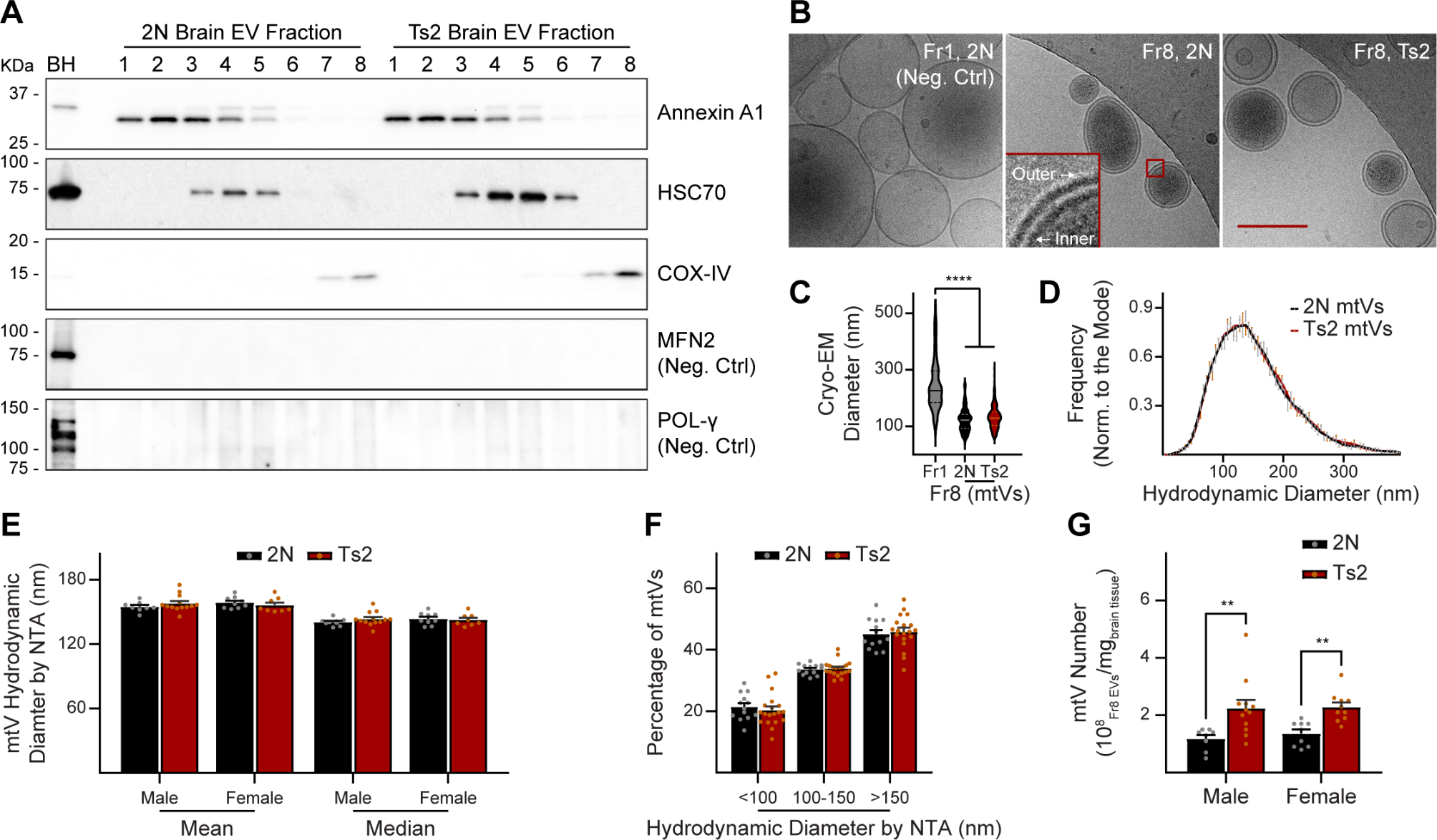

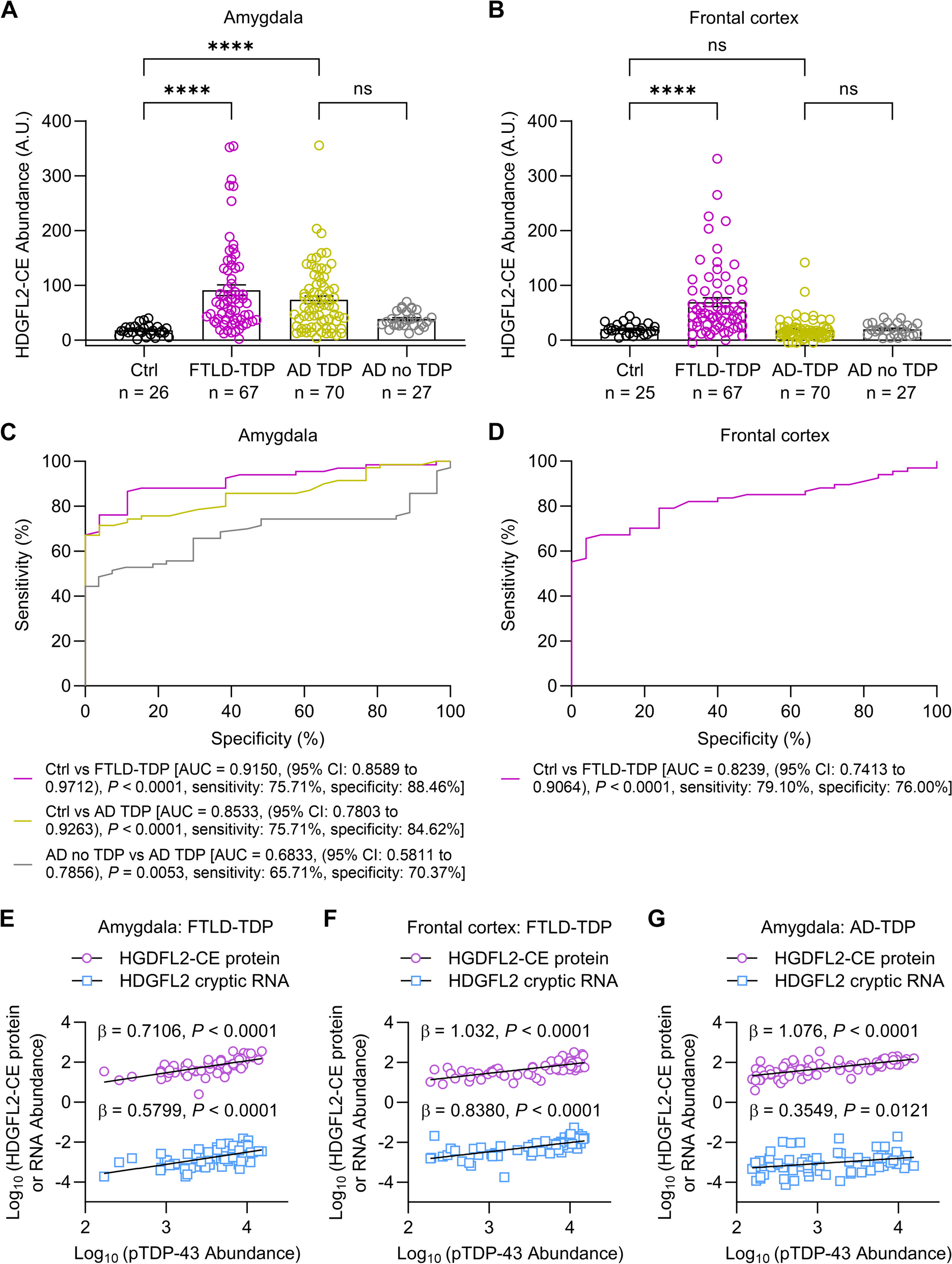
Tau is encoded by the microtubule associated protein tau (MAPT) gene, which is located on chromosome 17q21.3, containing 16 exons. In its precursor messenger RNA (pre-mRNA), alternative splicing of exon 2 (E2), E3 and E10 generates six tau isoforms, and the length range of tau protein is between 352 and 441 amino acids [1] (Fig. 1). Depending on the existence of three or four repeats of MTBDs (microtubule binding domains), tau can be classified into 3R tau (exon 10 exclusion) and 4R tau (exon 10 inclusion) [1, 3]. No isoform contains an isolated E3 without E2. The levels of the 3R and 4R isoforms are approximately equal in the adult human brain [4]. Another less discussed tau isoform is Big tau, which is also generated from alternative splicing of pre-mRNA from the MAPT gene. It has an extra E4a, leading to its higher molecular weight ~ 110 kDa. Due to its large size and few phosphorylated sites, Big tau was hypothesized to have a lower propensity to form pathological misfolding. This theory is in line with spared PNS (peripheral nervous system) in tauopathies, where Big tau is mainly expressed [5].
Fig. 1
MAPT pathological mutations in exons, haplotypes, and alternative splicing. MAPT (microtubule associated protein tau) gene, located on chromosome 17q21.3, has been identified with over 100 mutations, and pathological mutations associated with increased risk for tauopathies are shown above. The difference between H1 and H2 haplotypes is a 900 kb inversion existing in the largest linkage disequilibrium (LD) area in chromosome 17. Besides, H1 and its various sub-haplotypes usually contribute to disease occurrence, while H2 haplotype often act as a protective factor. Alternative splicing is common in neuronal cells which help to increase genetic plasticity and the diversity of proteome under physiological conditions [6]. However, imbalance in the ratio of the 3R and 4R isoforms can give rise to the pathogenesis of tauopathies, as the 4R tau is more efficient in promoting microtubule assembly with an extra repeat domain R2 which hyper-stabilize MT and more free-floating tau leads to aggregates formation [3, 6, 7]
Tau mainly exists in the axons of neurons under physiological conditions [1]. The MTBDs bind to tubulins, promoting microtubule assembly and stability. In addition, tau knock-out mice developed glucose intolerance, pancreatic disorders, anxiety, and impairment of contextual and cued fear memory, implying a wide range of undiscovered functions of tau [8]. Tau also plays a role in protecting genomic architecture, regulating myelination and synaptic plasticity, iron homeostasis, and neurogenesis [9, 10]. Further scrutiny might influence anti-tau therapies as the physiological functions of tau are unintentionally disturbed by treatment.
Regulation of affinity between tau and microtubules mainly depends on phosphorylation of tau, which mostly happens in proline-rich region and C-terminus [11]. However, hyperphosphorylation at some sites will reduce binding affinity, resulting in instability of microtubules, also affecting axonal transport and neurotransmission [11]. Besides, other alterations of tau such as overexpression, mutations, other aberrant posttranslational modifications in addition to phosphorylation (acetylation, truncation, O-GlcNAcylation, etc.), abnormal ratio between tau isoforms and mis-localization could lead to pathological changes observed in tauopathies [10].
The role of tau in neurodegeneration is still not elucidated. Mechanisms have been commonly summarized as loss of function, gain of function and mislocalization due to tau abnormality [10]. A recent study characterized tau interactomes in human induced pluripotent stem cell (iPSC)-derived neurons. They revealed the direct interactions between pathological tau and proteins, including SNARE complex, RNA-binding proteins, and mitochondrial proteins, which influenced tau release, protein synthesis and energy supply respectively. It helped to discover tau mediated pathogenesis and potential therapeutic strategies [12].
Epidemiology of tauopathiesAccording to WHO, more than 55.2 million people worldwide currently suffer from dementia, with approximately 7 million new cases per year, and the data will increase to 139 million in 2050 [13]. Aging is still the greatest risk factor of all. A cross-sectional study recruiting 46,011 adults over 60 in China suggested a significantly higher prevalence of dementia in population with older age (OR: 2.69–6.60), female gender (OR: 1.43) and family history (OR: 7.20) [14]. Other modifiable risk factors include education, overall health [15], lifestyle, rural residence and environmental factors [16] like PM 2.5 [17] and transportation noise [18].
Frontotemporal dementia (FTD) account for 2.6% of all-cause dementia. Though regarded as the most common presenile dementia, the incidence of FTD increased over time and peaked at the age of 75–79 [19, 20]. The age-standardized incidence of FTD was 2.90 per 100,000 person-year. Time ranges of survival changed slightly among bvFTD (behavioral variant of frontotemporal dementia) and AD, whereas FTD patients initially developing motor symptoms (all having PSP or CBD as underlying pathology) had the shortest survival time [19]. Accuracy of data about FTD is hampered by underdiagnosis and different criteria adopted. For better estimation, a multicenter prospective observational study designed in Europe is underway [21].
In Olmsted County, the incidence of PSP and CBS (corticobasal syndrome) increased over time, being 2.6 per 100,000 person-years and 0.4 per 100,000 person-years respectively [22], while another study found similar prevalence between them (around10.84/100,000) [23]. Most of patients are diagnosed at the age of 78 (70–74 for PSP, 75–79 for CBS [23]) with median survival time around 6 years [22].
AD is the most common type of dementia, accounting for 60–70% of all cases globally [19]. From 2010–2012, the prevalence was 14.7%, which remained steady since 1994 and was higher in low-income countries. However, the annual incidence dropped prominently from 2.8% in 1998–2000 to 2.2% in 2010–2012 [24]. Another study observed a 13% decrease per decade over the past 25 years [25]. Additionally, in non-western countries, the incidence of AD rose significantly among groups aged between 65 and 74 [26].
AGD is commonly regarded as a late on-set disease with mixed pathology. In a non-Caucasian population, 15.2% of participants were diagnosed with AGD, whose mean age were 78.9 ± 9.4 years [27]. In younger population with potentially purer AGD pathology, a study identified 7 patients via postmortem examination, only 2 were female. Most patients died at 64 years old with median survival time being 3 months (0.5–36) [28].
The clinical and neuropathological diagnosis criteria of CTE [29] haven’t reached a consensus due to the small number of cases studied. Thus, the exact incidence of CTE is unknown [30]. It has been reported that CTE is unavoidable in some sports. One study found that CTE pathology was more common in athletes and was mainly observed in men [31]. American football was the sport most associated with CTE (OR: 2.62) [31]. Longer playing years or a higher level of professionalism were linked to an increased risk of CTE and pathology severity [32, 33]. Prospective, autopsy studies of populations from different exposure backgrounds are currently needed to confirm its epidemiology and public impacts [30, 31].
Clinical symptomsTauopathies lead to a variety of behavioral, movement, language, and memory deficits [34, 35] (see in Table 1). Though we classify these symptoms into categories, overlap is common. In a cross-sectional study comprising 310 patients with frontotemporal lobar degeneration (FTLD), 62% of participants met the diagnostic criteria for more than one syndrome [36]. Such high continuity in phenotypes challenges the current mode, which diagnoses patients as discrete syndromes [36].
Table 1 Phenotypes of tauopathies and their salient featuresFTDFTD is an overall depiction of three clinical variants, including behavioral-variant frontotemporal dementia (bvFTD), non-fluent variant primary progressive aphasia (nfvPPA), and semantic-variant primary progressive aphasia (svPPA), which predominantly affect personality, behaviors, and language skills [37].
Overlap of three phenotypes is more prominent as disease deteriorates, and is consistent with pathology expansion to the large areas of frontal and temporal lobes. Motor disorder can also appear over time [36].
bvFTD can be diagnosed with neuropsychiatric symptoms and behavioral changes [38]. Criteria include a) early disinhibition (inappropriate manner and irritability), b) apathy, which is similar to depression, c) loss of empathy, d) early compulsive, repetitive, or stereotyped behaviors, impaired executive abilities and e) eating pattern changes including varied preference and hyperorality [38,39,40]. nfvPPA shows labored speech, sound errors, and impaired grammar but still retains single-word comprehension and plain semantics. To the contrary, svPPA manifests anomia, impaired comprehension of single words and object knowledge, though grammar and speech fluency are spared.
PSPSPSP syndromes (PSPS) encompass a range of behavioral, movement, and language disorders, among which PSP-RS (Richardson’s syndrome)—a classic movement disorder—is most widely studied. However, an autopsy based retrospective research indicated that up to 60–75% of patients with PSP pathology showed nonclassical variant PSP phenotypes which are outlined below, suggesting neglect in previous studies [41].
Postural instability and ensuing frequent falls are early signs of PSP-RS. But the diagnostic feature—vertical supranuclear ophthalmoplegia—usually develops in the 7th year after onset, or never appears. Patients also show mild dementia, dysarthria, dysphagia, executive dysfunction and non-levodapa responsive parkinsonism (axial, symmetric akinetic-rigid syndrome, and extensor neck dystonia) [34, 42,43,44]. After the appearance of clinical symptoms, people tend to live for 6.9 years on average [43].
PSP with predominant parkinsonism (PSP-P) is considered the second most common phenotype in PSPS followed by PSP with corticobasal syndrome (PSP-CBS) according to a prospective study [45]. PSP-P can be easily misdiagnosed as PD in initial stages for their akin manifestations. Retrospective diagnosis is made when they develop PSP-RS related manifestations.
In patients diagnosed with PSP, the symptom of progressive gait freezing (PSP-PGF) occurs as early as the onset of disease and deteriorates during illness [44]. It’s a pure movement deficit with no response to levodopa and can happen under specific circumstances or is a feature of PSP-RS [44].
PSP and CBS are clinically and genetically overlapping, as 44% of patients with CBS had PSP-like features and 30% of patients with PSP had CBS-like features [36]. PSP-CBS patients have PSP pathology and similar symptoms to CBS, including levodopa-resistant rigidity, bradykinesia, and apraxia. Clinical differentiation between PSP-CBS and CBD-CBS is thus impossible. When compared with PSP-RS, PSP-CBS has more severe ideomotor apraxia [45].
PSP-speech language (PSP-SL) phenotype depicts a syndrome which initially develops language disorders akin to nfvPPA before transformation into PSP-RS after years. PSP with frontal presentation (PSP-F) shows similar symptoms to bvFTD in early stages before developing motor dysfunctions like PSP-RS. Cerebellar ataxia appears at the onset of PSP with predominant cerebellar ataxia (PSP-C), but is not included in MDS PSP criteria [46].
CBSThe corticobasal syndrome (CBS) is characterized by asymmetrical limb apraxia, levodopa-resistant parkinsonism (dystonia, rigidity, tremor, and myoclonus) and deficits in higher cortical function [41, 47, 48]. Initial symptoms usually include cognition decline, meaning problems in vision, language, executive ability, and social cognition [47]. Another typical feature for CBS is alien/anarchic limb phenomena [49]. Other common symptoms include dysarthria and dysphagia.
Akin to PSP-RS, CBS shows equally severe abnormalities in movement involving limbs and eyeball [45]. Nonetheless, the description of freezing gait and myoclonus is lacking in the definition of PSP-RS, which are still the vital characteristics of CBS and PSP-PGF [45].
CTEApart from typical history of head injuries, CTE (chronic traumatic encephalopathy) is reported to have nonspecific symptoms including emotional disorders (depression, anxiety, irritability), bradykinesia, gait instability and cognition impairment. However, bias is inevitable as most studies were conducted retrospectively. Recently, the first prospective study comparing the clinical profiles of 6 CTE and 25 AD patients with autopsy evidence didn’t find a distinct clinical phenotype from AD, showing similar performance in motor, neuropsychological, behavioral, and cognitive evaluations [50].
ADSingle or multidomain amnesia (also named aMCI, amnestic mild cognitive impairment) is the most common manifestation in early stages of typical AD (Alzheimer’s disease) [37, 51], in which patients’ ability to form new episodic memories is damaged while other functions are conserved [37]. Some symptoms might occur long time before diagnosis including depression, sleep pattern changes, apathy and severe anxiety [51]. Typical symptoms are more common in LOAD (late onset AD) patients.
In atypical AD, other than memory loss, abnormalities in language, visuospatial dysfunction, impaired behavior, and executive abilities tend to appear earlier, and mostly happen in EOAD (early onset AD) population [37, 51]. Based on clinical features, atypical AD has been classified into three phenotypes, including logopenic variant primary progressive aphasia (lvPPA), which has anomia, fluency deficit, phonemic paraphasias and memory loss; posterior cortical atrophy (PCA), which has compromised advanced visual functions (visual field cuts, alexia, agnosia, ideomotor apraxia and cortically blindness) in early stages; and behavioral dysexecutive variant AD (bvAD), which can be misdiagnosed as bvFTD but shows milder behavioral dysfunction and earlier memory loss [37].
Pathology3R tauopathy
留言 (0)