Figure SI 1-1H NMR spectrum of compound 3a (CDCl3).
Figure SI 2-1H NMR spectrum of compound 3j (CDCl3).
Figure SI 3-1H NMR spectrum of compound 4a (CDCl3).
Figure SI 4-1H NMR spectrum of compound 4b (CDCl3).
Figure SI 5-1H NMR spectrum of compound 4c (CDCl3).
Figure SI 6-13C NMR spectrum of compound 4c (CDCl3).
Figure SI 7-1H NMR spectrum of compound 4d (CDCl3).
Figure SI 8-1H NMR spectrum of compound 4e (CDCl3).
Figure SI 9-13C NMR spectrum of compound 4e (CDCl3).
Figure SI 10-1H NMR spectrum of compound 5a (CDCl3).
Figure SI 11-1H NMR spectrum of compound 5b (CDCl3).
Figure SI 12-13C NMR spectrum of compound 5b (CDCl3).
Figure SI 13-1H NMR spectrum of compound 5c (CDCl3).
Figure SI 14-13C NMR spectrum of compound 5c (CDCl3).
Figure SI 15-1H NMR spectrum of compound 5d (CDCl3).
Figure SI 16-13C NMR spectrum of compound 5d (CDCl3).
Figure SI 17-1H NMR spectrum of compound 5e (CDCl3).
Figure SI 18-13C NMR spectrum of compound 5e (CDCl3).
Figure SI 19-1H NMR spectrum of compound 5f (CDCl3).
Figure SI 20-13C NMR spectrum of compound 5f (CDCl3).
Figure SI 21-1H NMR spectrum of compound 5 g (CDCl3).
Figure SI 22-13C NMR spectrum of compound 5 g (CDCl3).
Figure SI 23-1H NMR spectrum of compound 5 h (CDCl3).
Figure SI 24-13C NMR spectrum of compound 5 h (CDCl3).
Figure SI 25-1H NMR spectrum of compound 5i (CDCl3).
Figure SI 26-13C NMR spectrum of compound 5i (CDCl3).
Figure SI 27-1H NMR spectrum of compound 5j (CDCl3).
Figure SI 28-13C NMR spectrum of compound 5j (CDCl3).
Figure SI 29-1H NMR spectrum of compound 6a (CDCl3).
Figure SI 30-13C NMR spectrum of compound 6a (CDCl3).
Figure SI 31-1H NMR spectrum of compound 6b (CDCl3).
Figure SI 32-13C NMR spectrum of compound 6b (CDCl3).
Figure SI 33-1H NMR spectrum of compound 6c (CDCl3).
Figure SI 34-13C NMR spectrum of compound 6c (CDCl3).
Figure SI 35-1H NMR spectrum of compound 6d (CDCl3).
Figure SI 36-13C NMR spectrum of compound 6d (CDCl3).
Figure SI 37-1H NMR spectrum of compound 7a (CDCl3).
Figure SI 38-13C NMR spectrum of compound 7a (CDCl3).
Figure SI 39-1H NMR spectrum of compound 7b (CDCl3).
Figure SI 40-13C NMR spectrum of compound 7b (CDCl3).
Figure SI 41-1H NMR spectrum of compound 7c (CDCl3).
Figure SI 42-13C NMR spectrum of compound 7c (CDCl3).
Figure SI 43-1H NMR spectrum of compound 7d (CDCl3).
Figure SI 44-13C NMR spectrum of compound 7d (CDCl3).
Figure SI 45-1H NMR spectrum of compound 7e (CDCl3).
Figure SI 46-13C NMR spectrum of compound 7e (CDCl3).
Figure SI 47 – Chiral GC chromatogram for the alkylation of benzaldehyde, under optimized conditions with ligand 5i.
Figure SI 48 – Chiral GC chromatogram for the alkylation of 2-methoxybenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 49 – Chiral GC chromatogram for the alkylation of 3-methoxybenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 50 – Chiral GC chromatogram for the alkylation of 4-methoxybenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 51 – Chiral GC chromatogram for the alkylation of 2-methylbenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 52 – Chiral GC chromatogram for the alkylation of 3-methylbenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 53 – Chiral GC chromatogram for the alkylation of 2-chlorobenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 54 – Chiral GC chromatogram for the alkylation of 3-chlorobenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 55 – Chiral GC chromatogram for the alkylation of 4-chlorobenzaldehyde, under optimized conditions with ligand 5i.
Figure SI 56 – Chiral GC chromatogram for the alkylation of 1-naphthaldehyde, under optimized conditions with ligand 5i.
Figure SI 57 – Chiral GC chromatogram for the alkylation of 2-naphthaldehyde, under optimized conditions with ligand 5i.
Figure SI 58 – Chiral GC chromatogram for the alkylation of furfural, under optimized conditions with ligand 5i.
Figure SI 59 – Chiral GC chromatogram for the alkylation of cinnamaldehyde, under optimized conditions with ligand 5i.
Figure SI 60 – Chiral GC chromatogram for the alkylation of cyclohexanecarboxaldehyde, under optimized conditions with ligand 5i.
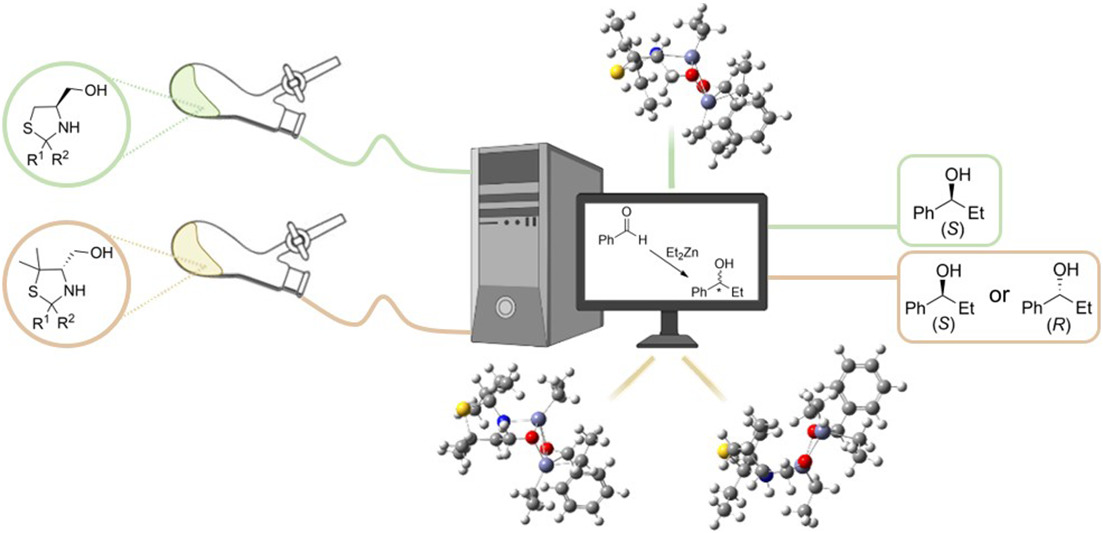
Figure SI 61 - Schematic representation of the TSS addressed in this study, in all possible configurations and conformations.
Figure SI 62 - Schematic representation of the PTSS addressed in this study, in all possible configurations and conformations.
Table SI 1 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-n-anti-(Re). Total energy: E = −4983.2710060057 Eh. Zero-point vibration energy: ZPE = 0.547322 Eh. Correction for Gibbs energy: Gcorr = 0.478894686726338 Eh.
Table SI 2 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-n-anti-(Si). Total energy: E = −4983.2787895164 Eh. Zero-point vibration energy: ZPE = 0.547925 Eh. Correction for Gibbs energy: Gcorr = 0.480289849552466 Eh.
Table SI 3 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-n-syn-(Re). Total energy: E = −4983.2766375209 Eh. Zero-point vibration energy: ZPE = 0.548378 Eh. Correction for Gibbs energy: Gcorr = 0.480460483717387 Eh.
Table SI 4 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-n-syn-(Si). Total energy: E = −4983.2676653321 Eh. Zero-point vibration energy: ZPE = 0.547694 Eh. Correction for Gibbs energy: Gcorr = 0.481733003237479 Eh.
Table SI 5 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-i-anti-(Re). Total energy: E = −4983.2784109434 Eh. Zero-point vibration energy: ZPE = 0.547478 Eh. Correction for Gibbs energy: Gcorr = 0.480479908588840 Eh.
Table SI 6 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-i-anti-(Si). Total energy: E = −4983.2805783057 Eh. Zero-point vibration energy: ZPE = 0.547816 Eh. Correction for Gibbs energy: Gcorr = 0.480526375928395 Eh.
Table SI 7 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-i-syn-(Re). Total energy: E = −4983.2801470555 Eh. Zero-point vibration energy: ZPE = 0.547624 Eh. Correction for Gibbs energy: Gcorr = 0.479478956389259 Eh.
Table SI 8 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 5i-PTSS-i-syn-(Si). Total energy: E = −4983.2801627503 Eh. Zero-point vibration energy: ZPE = 0.546914 Eh. Correction for Gibbs energy: Gcorr = 0.476946486383546 Eh.
Table SI 9 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-n-anti-(R). Total energy: E = −4983.2538242937 Eh. Zero-point vibration energy: ZPE = 0.546504 Eh. Correction for Gibbs energy: Gcorr = 0.481780613216530 Eh. Imaginary Frequency = −249.70 cm−1. C − C bond forming distance = 2.38 Å.
Table SI 10 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-n-anti-(S). Total energy: E = −4983.2599644880 Eh. Zero-point vibration energy: ZPE = 0.548381 Eh. Correction for Gibbs energy: Gcorr = 0.484070462768996 Eh. Imaginary Frequency = −243.97 cm−1. C − C bond forming distance = 2.34 Å.
Table SI 11 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-n-syn-(R). Total energy: E = −4983.2512919314 Eh. Zero-point vibration energy: ZPE = 0.547501 Eh. Correction for Gibbs energy: Gcorr = 0.482031993905923 Eh. Imaginary Frequency = −245.51 cm−1. C − C bond forming distance = 2.31 Å.
Table SI 12 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-n-syn-(S). Total energy: E = −4983.2314226410 Eh. Zero-point vibration energy: ZPE = 0.546611 Eh. Correction for Gibbs energy: Gcorr = 0.479402399542944 Eh. Imaginary Frequency = −527.45 cm−1. C − C bond forming distance = 2.33 Å.
Table SI 13 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-i-anti-(R). Total energy: E = −4983.2612540239 Eh. Zero-point vibration energy: ZPE = 0.547715 Eh. Correction for Gibbs energy: Gcorr = 0.484533993525043 Eh. Imaginary Frequency = −216.71 cm−1. C − C bond forming distance = 2.31 Å.
Table SI 14 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-i-anti-(S). Total energy: E = −4983.2568614281 Eh. Zero-point vibration energy: ZPE = 0.548064 Eh. Correction for Gibbs energy: Gcorr = 0.485515901733003 Eh. Imaginary Frequency = −277.30 cm−1. C − C bond forming distance = 2.39 Å.
Table SI 15 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-i-syn-(R). Total energy: E = −4983.2503281392 Eh. Zero-point vibration energy: ZPE = 0.547550 Eh. Correction for Gibbs energy: Gcorr = 0.483429442011045 Eh. Imaginary Frequency = −203.53 cm−1. C − C bond forming distance = 2.40 Å.
Table SI 16 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 5i-TSS-i-syn-(S). Total energy: E = −4983.2552489806 Eh. Zero-point vibration energy: ZPE = 0.547354 Eh. Correction for Gibbs energy: Gcorr = 0.482999428680251 Eh. Imaginary Frequency = −234.49 cm−1. C − C bond forming distance = 2.33 Å.
Table SI 17 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-n-anti-(Re). Total energy: E = −5061.8400954889 Eh. Zero-point vibration energy: ZPE = 0.603702 Eh. Correction for Gibbs energy: Gcorr = 0.533253094648638 Eh.
Table SI 18 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-n-anti-(Si). Total energy: E = −5061.8508433050 Eh. Zero-point vibration energy: ZPE = 0.603842 Eh. Correction for Gibbs energy: Gcorr = 0.532466577794706 Eh.
Table SI 19 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-n-syn-(Re). Total energy: E = −5061.8488122156 Eh. Zero-point vibration energy: ZPE = 0.60436 Eh. Correction for Gibbs energy: Gcorr = 0.535289659112550 Eh.
Table SI 20 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-n-syn-(Si). Total energy: E = −5061.8416182865 Eh. Zero-point vibration energy: ZPE = 0.60327 Eh. Correction for Gibbs energy: Gcorr = 0.532485240906494 Eh.
Table SI 21 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-i-anti-(Re). Total energy: E = −5061.8495831150 Eh. Zero-point vibration energy: ZPE = 0.603607 Eh. Correction for Gibbs energy: Gcorr = 0.533788992572843 Eh.
Table SI 22 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-i-anti-(Si). Total energy: E = −5061.8411270666 Eh. Zero-point vibration energy: ZPE = 0.603281 Eh. Correction for Gibbs energy: Gcorr = 0.533895638925919 Eh.
Table SI 23 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-i-syn-(Re). Total energy: E = −5061.8460169195 Eh. Zero-point vibration energy: ZPE = 0.603483 Eh. Correction for Gibbs energy: Gcorr = 0.533254237288136 Eh.
Table SI 24 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) optimization for 7d-PTSS-i-syn-(Si). Total energy: E = −5061.8422182559 Eh. Zero-point vibration energy: ZPE = 0.602325 Eh. Correction for Gibbs energy: Gcorr = 0.532946486383546 Eh.
Table SI 25 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-n-anti-(R). Total energy: E = −5061.8272382400 Eh. Zero-point vibration energy: ZPE = 0.604811 Eh. Correction for Gibbs energy: Gcorr = 0.540150828413635 Eh. Imaginary Frequency = −248.96 cm−1. C − C bond forming distance = 2.37 Å.
Table SI 26 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-n-anti-(S). Total energy: E = −5061.8347020097 Eh. Zero-point vibration energy: ZPE = 0.604547 Eh. Correction for Gibbs energy: Gcorr = 0.538835650352314 Eh. Imaginary Frequency = −240.38 cm−1. C − C bond forming distance = 2.31 Å.
Table SI 27 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-n-syn-(R). Total energy: E = −5061.8257999379 Eh. Zero-point vibration energy: ZPE = 0.603666 Eh. Correction for Gibbs energy: Gcorr = 0.535349076366406 Eh. Imaginary Frequency = −201.57 cm−1. C − C bond forming distance = 2.29 Å.
Table SI 28 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-n-syn-(S). Total energy: E = −5061.8213557867 Eh. Zero-point vibration energy: ZPE = 0.603107 Eh. Correction for Gibbs energy: Gcorr = 0.537539516282613 Eh. Imaginary Frequency = −250.40 cm−1. C − C bond forming distance = 2.36 Å.
Table SI 29 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-i-anti-(R). Total energy: E = −5061.8280497558 Eh. Zero-point vibration energy: ZPE = 0.603793 Eh. Correction for Gibbs energy: Gcorr = 0.537086269282042 Eh. Imaginary Frequency = −162.73 cm−1. C − C bond forming distance = 2.35 Å.
Table SI 30 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-i-anti-(S). Total energy: E = −5061.8231558330 Eh. Zero-point vibration energy: ZPE = 0.602725 Eh. Correction for Gibbs energy: Gcorr = 0.536007236716816 Eh. Imaginary Frequency = −246.40 cm−1. C − C bond forming distance = 2.37 Å.
Table SI 31 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-i-syn-(R). Total energy: E = −5061.8157510138 Eh. Zero-point vibration energy: ZPE = 0.602069 Eh. Correction for Gibbs energy: Gcorr = 0.534570177109122 Eh. Imaginary Frequency = −252.43 cm−1. C − C bond forming distance = 2.30 Å.
Table SI 32 - Cartesian coordinates (Å) obtained from the B3LYP/6-31G(d,p) transition state location for 7d-TSS-i-syn-(S). Total energy: E = −5061.8239382965 Eh. Zero-point vibration energy: ZPE = 0.603363 Eh. Correction for Gibbs energy: Gcorr = 0.537066844410588 Eh. Imaginary Frequency = −250.98 cm−1. C − C bond forming distance = 2.34 Å.
Figure SI 63-5i-PTSS-n-anti-(Re) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 64-5i-PTSS-n-anti-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 65-5i-PTSS-n-syn-(Re) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 66-5i-PTSS-n-syn-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 67 - PTSS-i-anti-(Re) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 68-5i-PTSS-i-anti-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 69 - PTSS-i-syn-(Re) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 70-5i-PTSS-i-syn-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 71-7d-PTSS-n-anti-(Re) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 72-7d-PTSS-n-anti-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 73-7d-PTSS-n-syn-(Re) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 74-7d-PTSS-n-syn-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 75-7d-PTSS-i-anti-(Re) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 76-7d-PTSS-i-anti-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 76-7d-PTSS-i-anti-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.
Figure SI 78-7d-PTSS-i-syn-(Si) optimized geometry at the B3LYP/6-31G(d,p) level of theory. Color code: grey, carbon; red, oxygen; blue, nitrogen; white, hydrogen; yellow, sulfur; violet, zinc.

留言 (0)