
記住我

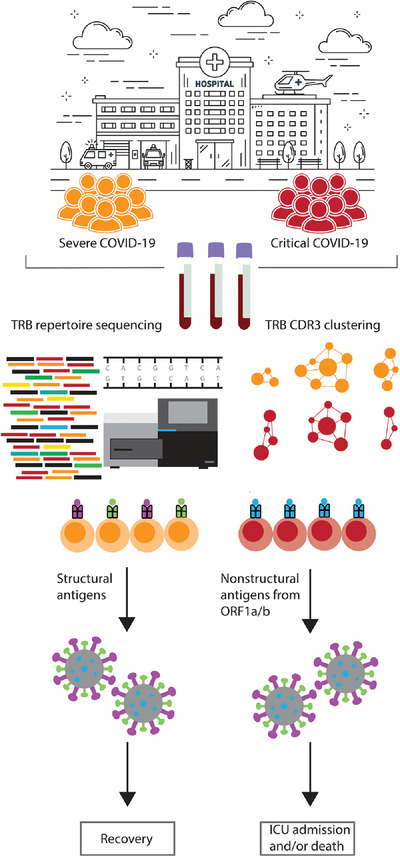
Clinical features of Coronavirus disease 2019 (COVID-19),1 caused by SARS-CoV-2 infection, can range from mild disease to severe illness with acute hypoxemic respiratory failure requiring intensive care unit treatment,2, 3 and potentially mechanical ventilation.4
The potential protective or pathogenic role of the adaptive immune response to SARS-CoV-2 has been vigorously debated. COVID-19 patients have been reported to consistently generate a substantial Tlymphocyte response against antigens derived from various SARS-CoV-2 proteins.5 Nevertheless, evidence of immune dysregulation in COVID-19 is accumulating, including reduced peripheral T-lymphocyte numbers, an exaggerated and/or prolonged inflammatory response, and T-lymphocytes displaying characteristics of exhaustion.6-8 The complexity of the growing evidence underlines the importance of the cellular immune response in COVID-19, alongside the established role of the humoral response.9-12
The T-cell receptor beta (TRB) repertoire is tremendously diverse. V-D-J recombination can theoretically result in the formation of 1012 TCRs, allowing recognition of essentially all peptide antigens presented by human leukocyte antigen (HLA) molecules.13 The TRB chain is most important in determining TCR diversity, the CDR3 region in particular is highly variable and heavily involved in binding to peptide HLA complexes. Accordingly, the CDR3 region of TRB sequences can be considered a unique barcode for T-lymphocytes targeting a particular antigen.14
Upon initiating a cellular immune response, T-lymphocytes undergo clonal expansion,15 which might be represented through a decreased diversity of the TRB gene repertoire and/or appearance of epitope-specific (e.g., COVID-19 specific) clonotypes.16 Clonotypes are mostly defined as sequences acquired by usage of the same V-, D-, and J-gene and identical CDR3 region, and can be readily identified by next-generation sequencing (NGS). However, it has been shown that epitope-specific TRBs often share conserved CDR3 features, without being fully identical. To this extent, epitope-specific TRBs often form clusters of clonotypes that share similar sequence features and therefore CDR3 regions.2
Until now, studies regarding the TRB repertoire in COVID-19 patients were largely descriptive17, 18 and a detailed analysis of the dynamics of the TRB repertoire in relation to disease course (i.e., severe vs. critical disease) has, to our knowledge, not been performed. We hypothesize that immunogenetic differences in the TRB repertoire may contribute to a severe (hospitalized) vs critical (ICU and/or death) disease course and that SARS-CoV-2 epitope-specific TRBs directed towards peptides from nonstructural proteins might be involved in the defective immune response that might lead to critical disease outcome. To test this hypothesis, we employed NGS of the TRB repertoire and characterized TRB sequences of interest which have highly similar TRB CDR3 amino acid sequences derived from TRB sequences enhanced in COVID-19 patients. These sequences were clustered using the TEIRESIAS algorithm. To determine which of the enhanced sequences in COVID-19 patients were SARS-CoV-2 epitope-specific, we utilized publicly available datasets of TRB-antigen associations.19, 20
2 RESULTS AND DISCUSSIONWe determined the temporal dynamics of the TRB repertoire in a total of 87 COVID-19 patients of whom 46 had a severe and 41 a critical disease course. No significant differences were observed in the cumulative frequency of the top 10 clonotypes (i.e., sum of frequencies of 10 most abundant clonotypes) in severe and critical COVID-19 patients at baseline and longitudinally (Figure 1A and B). Furthermore, the TRB Shannon diversity score did not differ between the severe and critical outcome groups (Figure 1C and D). Given the lack of overt differences in TRB repertoire between severe and critical COVID-19, we postulate that the T-lymphocyte response to COVID-19 may be mediated by several low frequency clonotypes, rather than by a single immuno-dominant clonotype. This idea is supported by other studies, where multiple smaller COVID-19 specific clonotypes were found in individual patients.20 Formally, we cannot entirely exclude that clonal T-lymphocytes have homed to the lungs and therefore may be underrepresented in the peripheral blood, as has been described in studies exploring paired TCR sequencing of bronchial alveolar lavage fluid and peripheral blood.21

Temporal dynamics of the top 10 clonotypes and Shannon diversity index in severe and critical COVID-19 patients. No significant differences were observed in the cumulative frequency of the top 10 clonotypes over time between patients with severe (A) and critical (B) disease course. Also, no significant differences were observed in Shannon diversity index of severe (C) and critical (D) COVID-19 patients. Red lines, derived from linear regression, represent the overall increasing or decreasing trend. The horizontal dashed lines represent the mean cumulative frequency of the top 10 clonotypes in 11 healthy controls, included as a reference. Severe group n = 46, Critical group n = 41.
The key to new insights based on the current dataset would be to establish which of the TRB clonotypes identified are specific for SARS-CoV-2. Public clonotypes, identical clonotypes shared across individuals, are a well-described phenomenon in both healthy and diseased subjects. In fact, mechanisms such as convergent recombination result in a far more limited TCR repertoire as theoretically possible. T-lymphocyte responses towards the same pathogen are often driven through the same antigens across individuals, resulting in epitope-specific clonotypes (enhanced sequences) which have been described for several viral diseases, including influenza, EBV, and CMV.2, 22 Therefore, we (and others) postulated that such enhanced sequences could be found in COVID-19 patients as well.23, 24 Sequences identified across individuals were considered shared if their CDR3 amino acid (AA) regions were at least 80% identical and 100% similar, meaning amino acids substitutions may only comprise of substitutions belonging to the same class. A cluster was defined as a collection of shared sequences that adhere to the aforementioned restrictions. The decision to study highly similar sequences rather than identical sequences was based on the assumption that CDR3 regions with only minor differences would still recognize the same antigen, especially when taking differences in HLA background into account, as previously reported.2, 25, 26
In our cohort, we identified 781 highly similar TRB CDR3 amino acid sequences representing 483 clusters in the 87 patients (Figure 2A). Most of the clusters (443) consisted of TRB sequences shared in 2 or 3 patients, with a minority (41) shared by ≥4 patients (Supplementary Table 1). We defined a cluster as “severe” or “critical” if all TRB sequences within that cluster were derived from patients with the same disease course. In total, we observed 96 severe clusters and 117 critical clusters, while the remaining 268 clusters were not associated with a particular outcome (Supplementary Table 1). To filter out shared public TRB sequences targeting common diseases such as EBV and CMV, we queried the VDJ database (VDJdb), an online repository for known TCR-antigen interactions.19 We discovered 54 of the 781 shared TRB sequences had previously been associated with an antigen from a disease other than COVID-19 in the VDJdb, suggesting that the prevalence of these TRB sequences was likely not related to COVID-19, albeit the potential for cross-reactivity cannot be completely excluded (Figure 2A).19
 Mapping of shared highly similar TRB CDR3 amino acid sequences to the SARS-CoV-2 genome. A Venn diagram of shared sequences between highly similar sequences identified in our cohort (TEIRESIAS), TRB sequences stored in the VDJ database of known antigen-TCR associations (VDJdb) and TRB sequences previously associated with SARS-CoV-2 antigens (MIRA) (A). Clusters associated with a critical disease course ((B), P P = 0.026), or NSPs encoded in ORF1a/b significantly (P SupplementaryTable 2b. Temporal dynamics of SARS-CoV-2-specific TRB sequences directed towards peptides from structural and nonstructural proteins in patients with severe (C) or critical (D) COVID-19 disease course. Severe group n = 46, Critical group n = 41. P-values are calculated using Fisher's Exact Test
Mapping of shared highly similar TRB CDR3 amino acid sequences to the SARS-CoV-2 genome. A Venn diagram of shared sequences between highly similar sequences identified in our cohort (TEIRESIAS), TRB sequences stored in the VDJ database of known antigen-TCR associations (VDJdb) and TRB sequences previously associated with SARS-CoV-2 antigens (MIRA) (A). Clusters associated with a critical disease course ((B), P P = 0.026), or NSPs encoded in ORF1a/b significantly (P SupplementaryTable 2b. Temporal dynamics of SARS-CoV-2-specific TRB sequences directed towards peptides from structural and nonstructural proteins in patients with severe (C) or critical (D) COVID-19 disease course. Severe group n = 46, Critical group n = 41. P-values are calculated using Fisher's Exact Test
As the remaining 727 shared TRB sequences were not associated with any antigens from previously studied diseases in the VDJdb, the prevalence of these TRB sequences in COVID-19 patients may be the result of stimulation by SARS-CoV-2 peptides. To further explore this, we utilized a dataset from a recently published study available through medRxiv that described the TRB repertoire of over 1500 COVID-19 patients and 3500 controls.20 The authors stimulated memory cells of 3 infected and 58 convalescent COVID-19 patients with SARS-CoV-2 specific peptide pools and sequenced SARS-CoV-2 specific CD8 T-lymphocytes through Multiplex Identification of T-cell Receptor Antigen Specificity (MIRA), thus generating a publicly accessible dataset with COVID-19 specific TCRs mapped to the SARS-CoV-2 genome.20, 27 Cross analysis of the TRB CDR3 amino acid sequences of our clusters with SARS-CoV-2 specific sequences from the MIRA-dataset revealed a match for 158 SARS-CoV-2 specific TRB sequences belonging to 134 clusters (Figure 2A; Supplementary Table 2a). These data indicate that our approach indeed identified SARS-CoV-2 specific TRB sequences and furthermore that there is overlap in the CD8 T-lymphocyte response to COVID-19 between Dutch, North-American and Italian patients. Until now, it remains unclear if any of the additional 569 highly similar TRB sequences, uniquely present in our dataset, are SARS-CoV-2 specific (Figure 2A).
Next, we selected the 113 SARS-CoV-2 specific clusters that exclusively targeted peptides from either structural or nonstructural proteins (NSPs) and screened for potential associations of clusters with disease course. We identified 19 clusters that were exclusively found in patients with a severe disease course, while 23 clusters were exclusively found in patients with a critical disease course (Supplementary Table 2b). Analysis of these clusters revealed a distinct skewing towards NSPs and accessory proteins of the SARS-CoV-2 genome for clusters associated with a critical disease course, while SARS-CoV-2 specific TRB sequences in clusters associated with severe disease course target peptides from structural proteins such as nucleocapsid phosphoprotein (N) and surface glycoprotein (S) (P = 0.026;Figure 2B). Upon further review, the skewing of the clusters associated with a critical disease course towards NSPs and accessory proteins appeared primarily mediated by the NSPs of ORF1a/b of the SARS-CoV-2 genome (P < 0.001) (Figure 2B; Supplementary Table 2b).
The observed skewing is interesting as multiple studies regarding SARS-CoV-1 have previously indicated that peptides from N and S proteins can induce strong SARS-CoV-1 specific T-lymphocyte responses.28-30 Additionally, the memory response to SARS-CoV-1 is almost exclusively directed towards peptides from N proteins.31 Furthermore, all candidate SARS-CoV-2 vaccines of major pharmaceutical companies are also primarily based on S proteins.32-34 Another study revealed that the vast majority of memory cells present in convalescent COVID-19 patients are directed toward peptides of the N protein, whereas almost none are directed toward peptides of NSPs.31 Even though peptides from NSP3 and NSP4 could induce a CD4 T-lymphocyte response in convalescent COVID-19 patients, these responses were an order of magnitude lower than responses towards peptides from N and S proteins.5 Furthermore, a recent publication reported that early induction of SARS-CoV-2-specific T-lymphocytes is present in patients with mild disease and accelerated viral clearance. The T-lymphocytes targeted primarily the N and S proteins, as well as the accessory proteins of ORF7/8.35 Altogether, our data indicate that an immune response to the nonstructural proteins of ORF1a/b of SARS-CoV-2 may be associated with an inability to sufficiently combat SARS-CoV-2 infection and consequently lead to a critical disease course.
We investigated the relationship between disease duration and the antigenic targets of the TRB repertoire (structural vs. NSPs of ORF1a/b). In the first 16 days after symptom onset, a consistent low frequency response of < 1% of the total TRB repertoire could be observed in 37 of 46 patients in the severe outcome group, primarily targeted at peptides derived from structural proteins (Figure 2C). For the patients with a critical outcome, a longer follow up was available, both due to closer monitoring in the ICU and due to prolonged hospital stay. We observed that clonotypes targeting peptides from nonstructural proteins of ORF1a/b are already present early during COVID-19 infection in patients with a critical outcome, in sharp contrast to the severe outcome group (Figure 2C and D). While clonotypes targeting structural antigens could be detected up to 24 days after symptom onset, all clonotypes detected after that point targeted peptides of NSPs of ORF1a/b (Figure 2D). Based on these observations, we hypothesize there may be some potential for clonotypes specific for peptides from nonstructural proteins of ORF1a/b in COVID-19 as a marker for critical disease.
Collectively our data illustrate a clear role for the T-lymphocyte response in COVID-19. Epitope-specific highly similar clonotypes can be found in the TRB repertoire of COVID-19 patients with both severe and critical disease course. We observed that SARS-CoV-2 specific TRB sequences in clusters associated with a critical disease course, targeted the nonstructural proteins of SARS-CoV-2 significantly more frequently than clusters associated with a severe disease course, which tended to target structural proteins. Therefore, T-lymphocyte reactivity towards peptides from NSPs of ORF1a/b may be related to a critical disease course. These results may be the first step towards new insights into the mechanisms behind COVID-19 disease severity, although our data need to be validated in larger cohorts that also include non-hospitalized COVID-19 patients with a mild disease course.
3 EXTENDED MATERIAL AND METHODS 3.1 PatientsEighty-seven patients with a positive SARS-CoV-2 PCR test and a clinically established COVID-19 diagnosis were included between March 24 and April 14, 2020, at the peripheral hospital Amphia Breda, the Netherlands. A detailed description of all individual patient characteristics is provided in Supplementary Table 3. Thirty-five patients (mean age 67, SD 8.6; mean BMI 28.5, SD 4.8) were admitted to the intensive care unit (ICU). Twenty of these patients were directly admitted to the ICU, while 15 were initially located at the inpatient clinic but were relocated to the ICU after a median admission time of two days (range 1 – 6). The other 52 patients (mean age 71, SD 10.7; n = 50 mean BMI 27.3, SD 4.9) were solely treated at the inpatient clinic. Six out of 35 ICU admitted patients (mean age 75, SD 7.1; mean BMI 27.1, SD 2.9) eventually succumbed after a median hospital admission period of 13 days (range 4 - 20) and a median ICU stay of 9.5 days (range 1.4–20.0). Four out of 6 of these patients (patients 5, 7, 8, and 16; Supplementary Table 3) had a history of underlying medical conditions while two (patients 4 and 13; Supplementary Table 3) did not suffer from any previous medical issues. Of note, however, 24 of 35 patients were still admitted to either the ICU (n = 21) or the inpatient clinic (n = 3) at the end of our study on May 19, 2020 (mean age 67, SD 8.2; mean BMI 29.0, SD 5.1). The other 5 patients (mean age 60, SD 6.7; mean BMI 27.9, SD 5.1) treated at the ICU recovered after a median ICU stay of 6.5 days (range 4.3–9.0) and a median post ICU stay at the inpatient clinic of 8 days (range 4–9). Six (mean age 82, SD 5.4; n = 5 mean BMI 24.9, SD 5.5) out of 52 non-ICU patients succumbed after a median hospital admission period of 7 days (range 4–10; patients 41, 55, 74, 77, 83, and 87; Supplementary Table 3) all these patients had signed an agreement stating that they did not wish to be transferred to the ICU or receive ventilation, which is a common procedure in the Netherlands. Forty-two patients recovered without ICU treatment (mean age 69, SD 10.0; BMI 27.6, SD 4.7) after a median hospital admission period of 7 days (range 2.0 – 13.0). Four of 52 patients solely treated at the inpatient clinic were still admitted at May 19, 2020, after a median hospitalization period of 47 days (range 46–52). Patients were divided into patients with a severe disease course (n = 46; i.e., patients admitted to the inpatient clinic) or critical disease course (n = 41; i.e., patients admitted to the ICU department and/or who succumbed to disease). Group averages are presented in Supplementary Table 4.
EDTA blood samples were collected from patients at multiple instances during the entire hospitalization period and occasionally after hospital discharge. See Supplementary Figure 1 for an overview of symptom onset, patient admission, discharge, disease course and sample collection. To study T-lymphocyte clonal expansion and repertoire during COVID-19, DNA was isolated from collected frozen peripheral EDTA blood samples and subsequently used for TRB repertoire analysis.
The study was performed in accordance with the guidelines for sharing of patient data of observational scientific research in case of exceptional health situations, as issued by the Commission on Codes of Conduct of the Foundation Federation of Dutch Medical Scientific Societies (https://www.federa.org/federa-english).
3.2 PCR amplification of TRBV-TRBD-TRBJ gene rearrangements and library preparationTotal cellular DNA was isolated from whole blood and 1000 ng was amplified through multiplex PCR of TRBV-TRBD-TRBJ gene rearrangements following the BIOMED-2 protocol.36 PCR products were gel-purified (QIAGEN, Hilden, Germany) and library preparation was performed according to the manufacturer's instructions (NEBNext® Ultra™ II DNA Library Prep Kit for Illumina®, NEB, Ipswich, MA, USA). The purity and size estimation of the libraries was assessed on an Agilent Bioanalyzer using the Agilent High Sensitivity DNA kit (Agilent Technologies, Lexington, MA, USA). The dsDNA HS Assay Kit was employed for quantification of the sequencing libraries on a Qubit 3.0 fluorometer (ThermoFisher Scientific, Waltham, MA, USA). Paired-end sequencing was performed using the MiSeq Reagent Kit v2 (2 × 250 bp) on the MiSeq Benchtop Sequencer (Illumina, San Diego, CA, USA). PhiX was spiked-in at a 20% concentration to increase library diversity.
3.3 NGS data bioinformatics analysisRaw FASTQ files were uploaded to the ARResT/Interrogate immunoprofiler for annotation and initial exploratory analysis.37 The full productive TRB repertoire for each sample was downloaded in .csv format and processed in R (R Core Team, 2020) during further analysis.38 Shannon diversity index was calculated using the vegan package.39 A summary of relevant QC metrics is depicted in Supplementary Table 5. For more details please refer to our entry in the GEO database under accession number GSE161810.
Clonotypes below 50 reads were excluded from clustering to reduce the influence of technical sequencing errors. Clustering was performed using the TEIRESIAS algorithm,24 a commonly used algorithm for pattern recognition in biological studies. The TEIRESIAS algorithm was initially developed by the Bioinformatics and Pattern Discovery group at the IBM Computational Biology Center23 and later adapted for CDR3 amino-acid sequences in chronic lymphocytic leukemia.24 We in turn adapted the identity and similarity thresholds to 80% and 100% respectively, to accommodate for the use of TCR data and to increase the likelihood that the identified highly similar TCRs indeed target the same epitope. Clustering was performed based on CDR3 amino acid sequence alone, TRBV and TRBJ gene identity was not taken into account for clustering.
The MIRA dataset was obtained from the ImmuneCODE data release.20 However, in this raw dataset of the MIRA results, 3178 out of 120,703 unique CDR3 amino acid TRB sequences targeted more than one peptide from the COVID-19 genome, with some TRB targeting as many as 15. As we deemed it unlikely that such promiscuous binding was entirely accurate, we applied an additional filtering step to these 3178 TRB sequences. In this filtering step, we discarded all peptide-TRB sequence associations that were reported in only one patient, while keeping the peptide-TRB associations reported in multiple patients, reducing this subset to 2647 TRB sequences with robust antigen associations. While this conservative approach certainly risks filtering out some true-positive hits, we considered it more important to prevent any doubts introduced by the potential false-positives in the MIRA-dataset. The 2647 TRB sequences and their peptide associations were merged back into the original MIRA dataset.
Both the MIRA dataset and our own clusters were screened for known TCR-antigen associations through the VDJdatabase (VDJdb) and matches were tagged.
The CDR3 amino acid sequences for each entry in the MIRA dataset were compared with the CDR3 amino acid sequences identified during clustering. All matches can be found in Supplementary Table 2a. For downstream analysis clusters matching with both the MIRA dataset and an unrelated target (autoantigen, other pathogen) in the VDJdb were filtered. Clusters with multiple targets within the same class (structural proteins vs nonstructural proteins) were grouped and labeled as targeting that class. Clusters not exclusively associated with an outcome or targeting peptides from both nonstructural and structural proteins were filtered.
3.4 Statistical analysisFrequency of a clonotype indicates the % reads of the total TRB repertoire in the patient sample. Cumulative frequency was calculated by summing the frequency of the 10 largest productive clonotypes in the repertoire. Cumulative frequency of the top 10 clonotypes and Shannon diversity score between outcome groups was compared through Wilcoxon signed-rank test. For the comparison of peptide targets between outcome groups, Fisher's Exact tests were applied on 2 × 2 contingency tables. All statistical analysis was performed in R (R Core Team, 2020).
ACKNOWLEDGEMENTSWe gratefully acknowledge Mr. P.J. Hengeveld for constructive discussion.
AUTHOR CONTRIBUTIONSB.S. and J.L.J.C. performed the experiments, interpreted results and wrote the manuscript, P.M.K. analyzed the data, interpreted results and wrote the manuscript; A.J.G., D.L. and T.A.A.M.E. collected the samples, interpreted results and helped write the manuscript; A.W.L., W.A.D. and V.H.J.V. designed and supervised the study, interpreted results and helped write the manuscript.
DISCLOSUREThe authors declare no conflict of interest.
Shared first author
Shared last author;
DATA SHARING STATEMENTThe data that support the findings of this study are available from the corresponding author upon reasonable request. TRB NGS data are uploaded in the GEO database and are available under accession number GSE161810.
ETHICAL STATEMENTThe study was performed in accordance with the guidelines for sharing of patient data of observational scientific research in case of exceptional health situations, as issued by the Commission on Codes of Conduct of the Foundation Federation of Dutch Medical Scientific Societies (https://www.federa.org/federa-english).
留言 (0)