
記住我







The mean prevalence of sensitization to birch pollen has been estimated to range from approximately 8% to 16% in general European populations, from 9.4% to 22% across different regions of Canada and from 11% to 20% across US regions.1-3 Pollen from birch and other members of the birch homologous group including alder, hornbeam, hazel, beech and oak is a major reason for allergic rhinitis and possibly also asthma symptoms.1, 4-6 The members of the birch homologous group are all characterized by containing allergen homologous to the major birch allergen Bet v 1.
Allergen immunotherapy (AIT) is the only available treatment modality with the potential to modify the natural course of the allergic disease by induction of tolerance.7 Recently, the SQ tree sublingual immunotherapy (SLIT)-tablet received regulatory approval across Canada, Europe and Switzerland for the treatment of tree pollen allergy induced by birch and other species in the birch homologous group. Hence, a detailed analysis of existing safety data obtained with this tablet in clinical trials will be of high interest to physicians and other healthcare providers within the field of allergy immunotherapy. The objective of this pooled safety analysis is therefore to provide detailed safety data for healthcare workers to be used in their clinical practice.
It has been estimated that SLIT is used in 45% of patients receiving allergen immunotherapy,7 and the safety profile of SLIT appears to be favourable when compared with subcutaneous immunotherapy (SCIT).7-10 Anaphylactic reactions occur with SLIT and SCIT products, but they are more frequently observed with SCIT products. This is why SLIT rather than SCIT products are recommended for at-home administration. Local application-site reactions occur commonly with both products,7 and the local application-site reactions with SLIT tend to be mild in nature, transient and self-resolving and occur most frequently early in the treatment period.9, 11
Around 70% of individuals with allergy to tree pollen from the birch homologous group also develop allergic symptoms against certain foods such as nuts and apples containing Bet v 1 homologous allergens, and the symptoms are manifested as a condition called pollen food syndrome (PFS).1, 12 The safety profile in individuals with PFS will be of special interest in the present safety analysis.
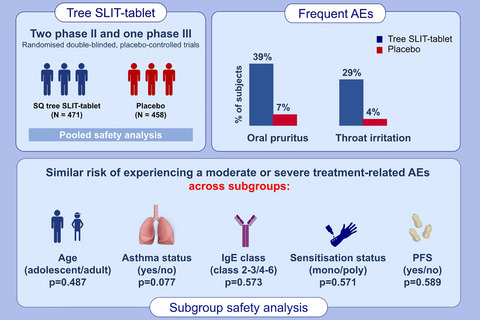
The clinical programme conducted so far with the SQ tree SLIT-tablet comprises one phase-I trial, two phase-II trials and one pivotal phase-III trial. All trials were conducted as randomized, parallel-group, double-blind, placebo-controlled clinical trials. This pooled safety analysis includes the two phase-II and the pivotal phase-III trials.
2 METHODSThe SQ tree SLIT-tablet is a fast dissolving lyophilisate for sublingual administration containing standardized allergen extract from birch pollen (Betula verrucosa). SQ-Bet is a measure of the biological allergen activity based equally on the major allergen content (Bet v 1) and total allergenic activity.
2.1 Trial designSafety data were pooled from two phase-II and one pivotal phase-III double-blinded, randomized placebo-controlled, parallel-group trials including adults and adolescents with moderate-to-severe allergic rhinitis and/or conjunctivitis induced by pollen from the birch homologous group. The data included subjects who received placebo or a daily dose of 12 SQ-Bet. The trials were conducted at 124 sites in total in Canada, Czech Republic, Denmark, France, Germany, Finland, Lithuania, the Netherlands, Norway, Poland, Russia and Sweden.
The subjects were treated before and during one pollen season with once-daily SQ tree SLIT-tablet or placebo administered at home. The first dose was administered under medical supervision for 30 min after tablet intake to assess tolerability and allow for possible treatment of any immediate side effects. The design as well as efficacy and safety results of the phase-II trials and the pivotal phase-III trial have been described separately elsewhere.13-15
2.2 PopulationThe study populations in the phase-II and phase-III trials comprised adolescents and adults (12–65 years) with persistent moderate-to-severe allergic rhinitis and/or conjunctivitis induced by birch pollen despite having received symptom-relieving medication during the 2 previous tree pollen seasons. Subjects had to have a positive skin prick test response (wheal diameter, >3 mm) to birch, a positive Bet v 1-specific IgE level (IgE class 2 or greater, >0.7 kU/L), and affected quality-of-life items (sleep disturbance; impairment of daily activities, leisure and/or sport; impairment of school or work; or troublesome symptoms) because of allergic rhinitis and/or conjunctivitis during the previous birch pollen season. Subjects with uncontrolled asthma or severe asthma exacerbations were excluded from the trial.
The subjects in the pooled safety analysis data set (hereafter called ‘pooled population’) comprised subjects from the mentioned phase-II and phase-III trials who were randomized to a daily dose of 12 SQ-Bet or placebo and received at least one dose of treatment.
2.3 End points and assessmentsA number of treatment-emergent adverse events (AEs) and AEs assessed by the investigator as possibly related to the investigational medicinal product (IMP-related) were summarized by treatment group and MedDRA System Organ Class (SOC), MedDRA Preferred Term and classified by severity (mild, moderate, severe), seriousness, action taken, time from first intake to AE (onset day) and reoccurrence after IMP administration. AEs were recorded from the point in time when subjects signed the informed consent form and up until the last follow-up. All AEs were coded according to the current MedDRA version at the time of trial conduct. For the pooled population, AEs were re-coded to MedDRA version 19.0.
The severity of an AE was a clinical observation assessed by the investigator using the following definitions:
Mild: Transient symptoms, no interference with the subject's daily activities Moderate: Marked symptoms, moderate interference with the subject's daily activities Severe: Considerable interference with the subject's daily activities, unacceptable.An AE was considered treatment-emergent if the time of onset was after the time of first IMP dose. However, if a subject discontinued, only AEs recorded up until 7 days after the discontinuation date were considered treatment-emergent.
2.4 Statistical methodologyIn addition to the pooled population, two other data pools were used in order to investigate the difference between adults and adolescents: One pool including adults (≥18 years) and one pool including adolescents (12– <18 years). The safety analyses were conducted according to the actual treatment that subjects received regardless of randomization.
Demographic (including age and gender) and baseline characteristics were summarized by treatment group. Onset and duration in days were summarized in subjects with or without medical history of PFS for the most frequent IMP-related AEs (≥5% of subjects in the 12 SQ-Bet treatment group). Onset is the time from randomization to the start of the AE. AEs were summarized in terms of treatment-emergent AEs and by causal relation to IMP, severity, seriousness and AEs leading to discontinuation (treatment-emergent AEs and AEs related to IMP, respectively).
A graphical overview was presented for the frequencies of the most frequent (≥5%) IMP-related AEs for subjects on active treatment in the pooled population by preferred term and worst case severity (ie., if a subject had more than 1 event, only the highest intensity was used). Likewise, IMP-related AEs in adults and adolescents with and without asthma were presented by treatment group (pooled population) and by worst case severity (for subjects treated with 12 SQ-Bet only).
To evaluate safety across additional intrinsic factors, an analysis of the differences between treatment groups in risk, calculated as hazard ratio, of experiencing first moderate or severe treatment-related AEs in specific subgroups (age (adolescents/adults), asthma status, IgE level (class 2–3 or class 4–6), sensitization (mono/poly) and PFS) was performed for the pooled population. The results were presented in a forest plot.
3 RESULTS 3.1 Subjects and baseline characteristicsThe pooled population comprised 929 randomized subjects (471 subjects treated with 12 SQ-Bet and 458 subjects treated with placebo) aged 12–65 years. Of the population exposed to 12 SQ-Bet, 35 subjects (7%) were adolescents. All randomized subjects received at least 1 dose of IMP. A total of 832 subjects (90%) completed the trials. Overall, the treatment groups appeared to be similar (Table 1).
TABLE 1. Demography and baseline characteristics of the pooled population Placebo (N = 458) 12 SQ-Bet (N = 471) Overall (N = 929) N %n N %n N %n Sex Male 220 (48%) 227 (48%) 447 (48%) Female 238 (52%) 244 (52%) 482 (52%) Age Mean (SD) 36.1 (13.2) 37.3 (13.4) 36.7 (13.3) Min–Max 12.0–65.0 12.0–65.0 12.0–65.0 SPT Birch only 8 (2%) 11 (2%) 19 (2%) Birch homologous group only 88 (19%) 99 (21%) 187 (20%) Birch homologous group only +others 369 (81%) 372 (79%) 741 (80%) Bet v 1 specific IgE class 2–3 226 (49%) 226 (48%) 452 (49%) 4–6 232 (51%) 243 (52%) 475 (51% Unknown - - 2 (<1%) 2 (<1%) Birch allergy Birch AR/C 458 (100%) 471 (100%) 929 (100%) Years with AR/C, mean (SD) 17.0 (11.8) 15.8 (10.8) 16.4 (11.3) Asthma (any cause) Asthma at baseline 168 (37%) 184 (39%) 352 (38%) Years with asthma, mean (SD) 12.9 (10.7) 12.0 (10.7) 12.4 (10.7) PFS PFS at baseline 296 (65%) 296 (63%) 592 (64%) Years with PFS, mean (SD) 14.9 (11.3) 14.2 (10.9) 14.5 (11.1) Note IgE class 2–3: Bet v1 IgE 0.71–17.5 kU/L; IgE class 4–6: >17.5 kU/L. Abbreviations: %, percentage of subjects in subgroup; N, Number of subjects in pool; n, number of subjects in subgroup; SD, standard deviation.Of subjects exposed to 12 SQ-Bet, 88% were exposed for 24–41 weeks, and 12% for less than 24 weeks. The average treatment duration of the three phase-II/III trials varied between 23 and 32 weeks.
3.2 Overall safetyA higher proportion of subjects reported IMP-related AEs with 12 SQ-Bet than with placebo (Table 2). IMP-related AEs were mainly local allergic reactions primarily in the oral cavity and throat and were related to the sublingual administration of IMP. They were mild or moderate in severity: 79% were mild and 17% moderate with 12 SQ-Bet and 65% were mild and 31% moderate with placebo. The majority resolved without treatment (76% of events for 12 SQ-Bet and 83% for placebo). No use of epinephrine was reported.
TABLE 2. Summary of AEs in the pooled population Placebo Placebo adolescents Placebo adults 12 SQ-Bet 12 SQ-Bet adolescents 12 SQ-Bet adults (N = 458) (N = 37) (N = 421) (N = 471) (N = 35) (N = 436) Subjects reporting N (%n) N (%n) N (n%) N (n%) N (%n) N (%n) Treatment-emergent AEs 289 (63%) 22 (59%) 267 (63%) 402 (85%) 29 (83%) 373 (86%) AEs related to IMP 144 (31%) 10 (27%) 134 (32%) 373 (79%) 28 (80%) 345 (79%) Severe treatment-emergent AEs 12 (3%) 1 (3%) 11 (3%) 39 (8%) 2 (6%) 37 (8%) Severe AEs related to IMP 2 (<1%) 1 (3%) 1 (<1%) 22 (5%) 1 (3%) 21 (5%) Serious treatment-emergent AEs 6 (1%) – – 6 (1%) 9 (2%) – – 9 (2%) Serious AEs related to IMP 1 (<1%) – – 1 (<1%) 1 (<1%) – – 1 (<1%) Treatment-emergent AEs leading to discontinuation 11 (2%) – – 11 (3%) 39 (8%) 1 (3%) 38 (9%) AEs related to IMP leading to discontinuation 8 (2%) – – 8 (2%) 33 (7%) 1 (3%) 32 (7%) Abbreviations: %n, percentage of subjects with events; N, number of subjects in pool; n, number of subjects with events.The risk of experiencing another AE was higher with moderate or severe AEs than with mild AEs. Among the most frequently reported AEs (>5% of subjects), around 50% of those who experienced a moderate or severe event had another AE of any severity (49.5 and 53%, respectively). For mild events, this rate was 36%.
3.3 Common IMP-related AEsThe most frequently reported IMP-related AEs with 12 SQ-Bet were oral pruritus (39% of subjects) and throat irritation (29%). This was the same for placebo but with a lower frequency: 7% (oral pruritus) and 4% (throat irritation) (Figure 1). Frequent IMP-related AEs outside the oral cavity related to treatment with 12 SQ-Bet were ear pruritus (13% of subjects), cough (6%) and sensation of foreign body (2%). Severe IMP-related AEs reported by more than 1 subject treated with 12 SQ-Bet were oral hypoaesthesia, oral paraesthesia, tongue pruritus, mouth swelling, cough, oropharyngeal pain and lip swelling.

Most frequently reported IMP-related AEs (>5%) for subjects treated with 12 SQ-Bet (pooled population) by severity
3.4 AEs leading to discontinuationThe majority of AEs leading to discontinuations were IMP-related: 33 subjects (7%) discontinued due to IMP-related AEs after treatment with 12 SQ-Bet and 8 subjects (2%) after receiving placebo. The most frequent IMP-related AEs leading to discontinuation after treatment with 12 SQ-Bet were throat irritation (10 subjects), oral pruritus (8 subjects), mouth swelling (8 subjects), swollen tongue (5 subjects), pharyngeal oedema (5 subjects) and ear pruritus (5 subjects). The majority of discontinuations due to IMP-related AEs occurred within the first few weeks of treatment. There were no differences between PFS subgroups in discontinuations due to AEs. However, for subjects treated with 12 SQ-Bet, a greater proportion of subjects had IMP interruptions in the PFS subgroup (19%) compared to subjects with no-PFS (7%).
3.5 Onset and duration of common IMP-related AEsThe most frequently reported IMP-related AEs had onset early during treatment, and the median time of onset was within the first week of treatment (Figure 2). The local allergic reactions reported most frequently such as oral pruritus, throat irritation, tongue pruritus, paraesthesia oral and pharyngeal oedema all had median onset on day 1. Very few subjects had onset of new events after two weeks of treatment.
 Median onset (•) and duration (−) of most frequently reported IMP-related AEs (>5%), displayed by subjects’ medical history of pollen food syndrome (PFS; N, no; Y, yes) and number of AEs for subjects treated with 12 SQ-Bet (pooled population).
Median onset (•) and duration (−) of most frequently reported IMP-related AEs (>5%), displayed by subjects’ medical history of pollen food syndrome (PFS; N, no; Y, yes) and number of AEs for subjects treated with 12 SQ-Bet (pooled population).  : Subjects without PFS (n = 175):
: Subjects without PFS (n = 175):  : Subjects with PFS (n = 296)
: Subjects with PFS (n = 296)
Median duration of the AEs was less than two weeks for most types of the most frequent IMP-related AEs (Figure 2). When considering the difference between those with and without PFS, no clear conclusions can be drawn in relation to onset and duration (Figure 2). In the group treated with 12 SQ-Bet, the longest median durations of most frequent IMP-related AEs were seen with dyspepsia (75 days), dry mouth (25 days), oral pain (21 days) and nasal pruritus (18 days) for all subjects (with and without PFS).
3.6 Serious AEsNo deaths were reported in the pooled phase-II/III trials. 15 subjects (2%) experienced 15 SAEs, 9 of these were reported with 12 SQ-Bet and 6 with placebo. 2 SAEs were assessed as IMP-related: 1 in each of the two groups, both involving accidental intake of IMP by subject's child. Both cases were asymptomatic as has previously been described.13
3.7 Safety in special groupsNo events of anaphylactic reactions or eosinophilic esophagitis (EoE) were reported. A subgroup analysis did not show any major differences in the risk of experiencing first moderate-to-severe IMP-related AEs across subgroups including age, asthma history, sensitization, IgE level and PFS status (Figure 3). Across all subgroups, hazard ratios were less than 1 indicating an increased risk of IMP-related AEs in the active treatment group compared with placebo as expected. The subgroup analysis did not show any major differences in the risk of experiencing a moderate or severe IMP-related AE across subgroups, including age (adolescents versus adults, p = .487), sensitization status (mono- vs. poly-sensitization, p = .571), birch IgE class (class 2–3 vs. 4–6, p = .573), asthma status (asthma vs. no-asthma, p = .077) and PFS status (PFS vs. no-PFS, p = .589).

Forest plot of hazard ratios of first moderate or severe IMP-related AE in various subgroups
3.7.1 PFSAt baseline, 296 (63%) and 296 (65%) of subjects treated with 12 SQ-Bet and placebo, respectively, reported to have PFS. After treatment, 9 subjects reported AEs of PFS as a specific response to food intake: 7 subjects treated with 12 SQ-Bet and 2 subjects treated with placebo. All AEs were assessed as IMP-related when treated with 12 SQ-Bet and 1 was assessed as IMP-related with placebo. Since 5 of 7 subjects reporting the PT oral allergy syndrome in the 12 SQ-Bet group also had a medical history of PFS, the data suggest that PFS may worsen during initiation of treatment with the SQ tree SLIT-tablet.
A larger proportion of subjects who reported PFS at baseline reported AEs compared with those without PFS. This was observed for both treatments: 89% of subjects with PFS and 80% without PFS experienced AEs with 12 SQ-Bet and 67% versus 56% experienced AEs with placebo. Likewise, of those treated with 12 SQ-Bet, oral pruritus was reported by 45% of subjects with PFS and by 29% of subject with no PFS. No major differences between subjects with and without PFS were seen for severity of AEs. A greater proportion of subjects had IMP interruptions in the PFS subgroup (19%) compared with subjects without PFS (7%). Most frequent treatment-emergent AEs (≥5% of subjects treated with 12 SQ-Bet), according to PFS status, can be found in the online supporting information (Table S1).
3.7.2 AsthmaAsthma was registered in the subjects’ medical history recorded at trial inclusion as any cause of asthma (Table 1). During the study, the 12 SQ-Bet treatment did not seem to induce an increased risk of asthma events. In total, 18 AEs of asthma were reported (7 in 7 subjects treated with 12 SQ-Bet and 11 AEs in 10 subjects treated with placebo). 6 AEs out of 18 were asthma exacerbation: 2 in 2 subjects treated with 12 SQ-Bet and 4 in 3 subjects on placebo. The majority of asthma AEs were mild or moderate in severity and assessed as unlikely related to IMP.
No differences were found between adult subjects with and without asthma in their medical history in relation to the percentage of subjects reporting IMP-related AEs with 12 SQ-Bet (Figure 4A). For subjects treated with 12 SQ-Bet, the proportion of adult subjects who reported any severe IMP-related AEs was 7% in subjects with asthma registered in their medical history at inclusion compared with 3% for subjects with no asthma at inclusion (Figure 4B). Slightly more adolescents with asthma in their medical history tended to experience AEs than adolescents without asthma (85% vs. 77%), and these AEs were mainly mild in severity. However, these data should be interpreted with caution as only 13 adolescent subjects with asthma medical history participated in the clinical phase-II/III trial programme.

A IMP-related AEs in adults and adolescents with and without asthma (pooled population) by treatment group. B IMP-related AEs by severity in adults and adolescents with and without asthma for subjects treated with 12 SQ-Bet
3.7.3 EczemaSeven subjects had treatment-related events of eczema (4 subjects treated with 12 SQ-Bet and 3 subjects treated with placebo). Three of the subjects treated with 12 SQ-Bet had no medical history of eczema and received no AE treatment for the event. For one subject treated with 12 SQ-Bet, the event was reported as a worsening of pre-existing atopic dermatitis: worsening of eczema periocular, perioral, and of the neck and the arms, including appearance of the ‘anguli infectiosi’. This subject was treated with topical corticosteroid and discontinued treatment with 12 SQ-Bet.
3.7.4 UrticariaUrticaria related to the IMP was reported by 6 subjects treated with 12 SQ-Bet and 4 treated with placebo. The 6 subjects treated with 12 SQ-Bet experienced contact urticaria at the administration site (2 subjects reported lip urticaria and 1 subject reported local urticaria), urticaria at the hand (1 subject), urticaria in several places of the body (1 subject reported urticaria on the neck after 3 days and on the chest and upper arm after 212 days) and for 1 subject no details were specified. 6 subjects in the placebo group also reported urticaria. In 4 of the subjects, the events (5 events) were assessed as possibly related to the treatment despite the subject never being treated with the tree SLIT-tablet. The events included 4 local reactions and 1 with unspecified location. In the remaining 2 subjects, urticaria was assessed as unlikely related to the tree SLIT-tablet.
This suggests either a systemic urticaria or a local urticaria resulting from touching the SLIT-tablet. None of the urticaria events were severe or lead to discontinuation.
3.8 Safety in adolescentsNo major differences were observed between adolescents (12–17 years) and adults (≥18 years) in the proportion of subjects reporting AEs including causality, severity, seriousness and discontinuation (Table 2). Also, for IMP-related AEs, no difference was observed, indicating no difference in the overall safety and tolerability between the two subgroups (Table 2).
4 DISCUSSIONThese results from a pooled safety analysis of clinical trials with 929 adults and adolescents with allergic rhinitis and/or conjunctivitis induced by birch pollen revealed no major safety concerns in relation to daily at-home use of the SQ tree SLIT-tablet. Thus, the safety profile supports daily at-home sublingual administration once the first dose is tolerated when administered under medical supervision: Most application-site reactions were local reactions of the mouth, throat and ear and were generally occurring early in treatment and were transient and self-resolving. Also, no anaphylactic reactions or IMP-related SAEs were reported for the subjects.
The findings are in accordance with the findings of systematic reviews: The most commonly reported adverse reactions with SLIT products are local adverse reactions primarily involving the oropharyngeal regions, severe systemic reactions are rare, and the reactions tend to be local and mild in nature and occur in the beginning of the treatment.7, 16
The results confirm previous safety findings with other SQ sublingual immunotherapy tablets. A grass SLIT-tablet showed a similar safety profile dominated by local allergic application-site reactions primarily in the oral cavity and throat of mild or moderate severity with an early onset and transient and self-resolving AEs.17, 18 Also, the safety profile of a house dust mite SLIT-tablet revealed transient, mild local allergic reactions representing the most commonly occurring adverse reactions.19
Around 70% of individuals with birch pollen allergy develop pollen food syndrome (PFS),1 and in the present study, 64% of subjects had PFS. This PFS subpopulation could be expected to be more challenged in relation to their allergic disease, and the safety profile may therefore differ compared with a subpopulation with no PFS.20 In the present data, subjects with PFS tended to experience more AEs regardless of treatment than subjects without PFS, especially oral pruritus. Of subjects treated with 12 SQ-Bet, oral pruritus was reported by almost half of the subjects with PFS (45%) and was thus far more frequent than in subjects without PFS (29%). This difference was not seen in relation to severity. This high reporting of oral pruritus is in accordance with what is usually seen in relation to PFS.20
The present data suggest no increased risk of asthma events among all subjects treated with 12 SQ-Bet. In addition, no acute asthma worsening was seen in subjects with a medical history of asthma. This is in line with the general safety profile of sublingual immunotherapy revealing no greater risk for adverse reactions in well controlled asthma.7 However, in the studied population, subjects with severe and/or uncontrolled asthma were excluded, and the safety profile of 12 SQ-Bet remains to be investigated in this subpopulation.
Patients with birch pollen allergy may show urticaria or worsening of atopic dermatitis when exposed to birch pollen.21, 22 In the present pooled safety analysis, only one event of worsening of atopic dermatitis was reported; however, we saw more events of urticaria in the group treated with 12 SQ-Bet compared with placebo. Contact urticaria following direct contact to allergen containing SLIT tablets are to be interpreted like PFS developing in the mouth. Only one subject in the
留言 (0)