
記住我








Diabetic kidney disease (DKD) is a serious public health problem and is the leading cause of end-stage kidney disease (ESKD) worldwide1. It is a significant complication of both type 1 diabetes mellitus and type 2 diabetes mellitus. DKD is the leading cause of type 2 diabetes, with approximately 30–50% of cases attributable to diabetes1-3. The growing incidence of type 2 diabetes contributes to the increasing incidence of DKD, which is associated with increased cardiovascular complications, morbidity and mortality4-7.
The mechanisms leading to the development and progression of renal injury in diabetes are not fully understood. Confounding factors have been reported to be associated with the pathophysiology of DKD, including absolute or relative insulin deficiency8, oxidative stress9, activation of the renin–angiotensin–aldosterone system10, formation of polyol and advanced glycation end-products11, hemodynamic alterations12, and variations in deoxyribonucleic acid methylation profiles13. The presence of proteinuria, along with elevated levels of glycated hemoglobin, systolic blood pressure and serum uric acid, and the presence of vascular comorbidities, are indicators of a faster progression of DKD14. Many individuals with DKD develop albuminuria15. The remission of albuminuria and a slowing down of the rate of loss of renal function might occur with optimization of glucose management and blood pressure control, including the use of renin–angiotensin system blockade, sodium–glucose cotransporter 2 inhibitors and glucagon-like peptide-1 receptor analogs4, 10, 16. However, despite these therapeutic options, DKD remains a major public health issue, not only due to the increased risk of progression to ESKD, especially in an aging population, but also because people with DKD are at a much higher risk of developing cardiovascular disease17-20. It is therefore crucial to understand the underlying mechanisms of DKD and to target these mechanisms to prevent or treat DKD.
The risk of developing DKD is associated with both systemic and the local activation of inflammatory processes21. Such inflammation can be triggered by stimulated renal cells and the accumulation of immune cells from both the innate and adaptive immune responses within the injured kidney, driving remodeling of renal structure and interstitial fibrosis22. Macrophages are phagocytic cells of the innate immune system, and are widely recognized to hasten the progression of glomerular sclerosis in experimental DKD models and clinical trials23-25. More than 90% of leukocyte infiltration in the kidney are macrophages23, 26, and strategies to selectively reduce macrophage accumulation, such as knockout of macrophage chemokines or blockade of chemokine receptors, provide significant protection in mouse DKD models27, 28. Growing evidence from experimental and clinical studies show that the adaptive immune system in concert with several inflammatory cytokines might also act as a key factor in the progression of renal injury in diabetes22, 29, 30.
The adaptive immune system comprises T cells and B cells. The progression of human DKD correlates with activation of T cells in the blood and increased numbers of CD4+ T cells in the kidney21, 31. CD4+ T-cell subsets have been intensively studied in DKD, including T helper (Th) 1, Th2, Th9, Th17, Th22, T regulatory cells (Tregs) and follicular helper T cells32. Th1 cells, but not Th2 cells, produce large amounts of cytokines, which are associated with lower levels of creatinine clearance and increasing proteinuria in people with diabetes and nephrotic syndrome (urine protein loss >3.5 g/day), as compared with those having diabetes without nephrotic syndrome33. The ratios of CD4 + CD25hi Tregs/Th17 cells and CD4 + CD25hi Tregs/Th1 cells are decreased in the blood of people with type 2 diabetes with reduced immunosuppressive potentiality of CD4 + CD25hi Tregs34. Further evaluation showed no correlation between this ratio and glycated hemoglobin in people with type 2 diabetes. Similarly, the Tregs/Th17 balance is disturbed in diabetes, with the percentage of Th17 cells being significantly higher and level of Tregs being lower in the blood of people with type 1 diabetes compared with healthy controls, which might exacerbate diabetic microvascular complications35, 36. Therefore, identifying the distinct CD4+ T-cell subset polarization and pathways involved in the dysregulation of the immune balance in DKD is extremely necessary for prognostication and treatment purposes.
Accompanying the invasion of leukocytes (such as neutrophils and macrophages) from the innate immune system, autoreactive T cells and B cells promote the production of autoantibodies, which target type IV collagen in the glomerulus (such as anti-glomerular basement membrane glomerulonephritis [anti-GBM GN]), or just accumulated as immunocomplexes in the glomeruli (such as a certain type of anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and immunoglobulin A nephropathy)37. In a murine model of Adriamycin nephropathy (chemical-induced-nephropathy), a model of focal segmental glomerulosclerosis, macrophages modified ex vivo by interleukin (IL)-10/transforming growth factor (TGF)-β were able to inhibit CD4+ T-cell proliferation and did not promote fibrosis in inflamed kidney38. In another mice model of crescentic glomerulonephritis (anti-GBM GN), T cells residing in the interstitium of the kidney showed the Th1 phenotype, and produced interferon-γ (IFN-γ)39 – a key factor of stimulating macrophages into the active state, as indeed presented by a pronounced infiltration of proliferating macrophages in the kidney of rats in early studies40-42. These data highlight the importance of further study on the interaction between T cells and other immune cells, such as whether T cells precede macrophages in the development and progression of kidney disease, as well as in DKD.
PATHOGENIC ROLE OF T CELLS IN THE DEVELOPMENT OF DKD Activation of T cells under the hyperglycemia milieu and other factors in DKDStudies have found enhanced levels of T-cells activation and proliferations linked to hyperglycemia43-46. A recent study found reduced capacity of T-cells proliferation in diabetic INSC94Y transgenic pigs (a large animal model showing a permanent diabetes phenotype after birth) compared with wild-type littermates47. In that study, proteomic analysis showed a high abundance of pathways associated with the immune system, signal transduction and metabolic function. Of the most regulated pathways, lipophagy is of particular interest as it involves with metabolic dysfunction of immune cells48, which suggests an altered metabolic phenotype of immune cells under the diabetic microenvironment.
Pathogenic role of T cells on induction of albuminuria in DKDSeveral studies suggest a pathogenic role of T cells for the induction of proteinuria during the development of DKD21, 31, 49. In people with type 2 diabetes, interstitial infiltration of CD4+ T cells correlated with the degree of proteinuria21. Similarly, in the blood of people with type 1 diabetes, the absolute number and percentage of T cells were significantly increased in patients with non-nephrotic proteinuria (>0.5 g and <3.5g/24 h) compared with controls (<0.5 g/24 h)31. Animal models of diabetes enhanced the pathogenic role of T and B cells in inducing albuminuria in DKD. In one study, Rag1−/− diabetic mice models lacking mature T and B cells showed protection against increasing albuminuria, which was characterised by slower progression of urine albumin excretion rate and albumin/creatinine ratio compared to wild-type control mice, implying that the absence of T and B cells lessened the risk of developing albuminuria29. Furthermore, it also showed that a systematic inhibition of T-cell activation by the drug, abatacept, ameliorated the presence of albuminuria, even when albuminuria was established in a streptozotocin (STZ)-induced type 1 diabetes model50. Notably, subsequent data showed no change of B7-1 (an immune-related protein that can be expressed by podocytes and leads to podocyte destruction in DKD51) after abatacept treatment in kidneys of diabetic mice compared with non-STZ mice, neither in cultured human podocytes nor in glomeruli of people with diabtetes50. Therefore, the protective effect of abatacept was thought to be predominantly by inactivating systematic T cells, rather than interacting with podocytes. These findings suggest that the increase of T or B cells systematically and locally contributes to the immunopathological process of the development of proteinuria in DKD.
Podocytes play an integral role in maintaining the glomerular filtration barrier integrity52, 53, where proteinuria occurs when compromised52, 54. One clinical study using 37 biopsies from Pima people with type 2 diabetes showed that podocyte detachment was significantly higher in people with macroalbuminuria (1.48%) than those with normal albuminuria (0.41%) or microalbuminuria (0.37%)55, confirming the same positive relationship between podocyte numbers and albuminuria, as shown in people with type 1 diabetes56. Notably, podocytes detachment, rather than podocytes numbers, showed a positive association with albuminuria in that study. This evidence indicates that podocyte dysfunction, instead of podocyte loss, might be a prime driver of impaired permselectivity in the glomerulus. Although DKD pathogenesis theories focused on podocyte dysfunction for a long time, the underlying mechanism contributing to the initial injury of podocytes has not been fully identified57. A recent study used ovalbumin (OVA) as a model antigen together with transgenic OT-I T cells bearing a T-cell receptor specific for OVA257–264 (SIINFEKL)58. Using this model, SIINFEKL peptide promoted OT-I T-cell proliferation and secretion of diabetes associated-inflammatory cytokines, such as IFN-γ and IL-17, and in turn aggravated podocyte injury and apoptosis59, 60. This finding suggests that T cells and podocytes act synergistically in the profibrotic progression of DKD.
TISSUE-RESIDENT MEMORY T CELLS IN INFLAMMATORY DISEASE: POTENTIAL TARGETS FOR THERAPY Ontogeny of tissue-resident memory T cellsAfter a pathogen encounter, there is a rapid production of effector T cells from naïve T cells that migrate swiftly to lymphoid and non-lymphoid tissues61. After infection resolution, antigen-specific effector T cells might differentiate into various memory T-cell subsets with distinct trafficking properties62. Effector memory T cells can circulate through lymphoid and non-lymphoid organs, whereas central memory T cells recirculate between lymph nodes, lymph and blood by the vascular addressin L-selectin (CD62L) and chemokine receptor CCR761, 63. In addition, a non-recirculating T-cell population persists in barrier tissues, including the skin, reproductive tract, respiratory tract, salivary glands and non-barrier tissues, including the brain and kidney63-68. These cells that exist in disequilibrium from the circulation are termed tissue-resident memory T (Trm) cells69-71 (Figure 1).

T-cell migration paradigm. Memory T cells are divided into circulating and non-circulating subsets. Central memory T (Tcm) cells migration is similar to that of the naïve T cells, and these cells predominantly reside in lymphoid tissues. Effector memory T (Tem) cells can pass through lymphoid and non-lymphoid organs, and join the blood through lymphatic vessels. Tissue-resident memory (Trm) cells are positioned within tissues and do not recirculate during steady-state conditions. NLT, non-lymphoid tissue; SLO, secondary lymphoid organs.
Prime markers of Trm cells in the human kidneyThe location where the effector T cells differentiation occurs is important for shaping the phenotypic expression of Trm cells61. Trm cells adopt a pattern of surface molecules lacking lymph node homing molecules CCR7 and CD62L, which set it apart from both central memory T and effector memory T cells61, 72. CD103+ Trm cells among CD8+ T-cell subsets were widely detected in the small intestine, skin epidermis, sensory ganglia and brain70, 73, and CD69 was widely expressed on the cell surface of Trm cells, so the co-expression of CD103 and CD69 was commonly used as the existence of Trm cells in many studies63, 74-76. However, the co-expression of CD103 and CD69 as markers for Trm cells has limitations, and a comprehensive analysis focusing on the migrational properties of T cells found that T cells lacking CD103 or CD69 can also be resident within the pancreas, salivary glands and female reproductive tract (FRT)77. With regard to the kidney, one study showed that a subset of CD8+ T cells displayed with a Trm-like phenotype (CD62L-CD69+CD103+) in the kidney of mice infected with lymphocytic choriomeningitis virus Armstrong64. However, it is worthy to note that a substantial proportion of CD8+ T cells in the mouse kidney lack expression of CD103, in agreement with those found in healthy human kidney biopsies78. Additionally, CD69 is also a marker of recent T-cell activation and is expressed on T cells at sites of chronic inflammation79, 80. Therefore, these results raise caveats that we need to interpret with caution, as Trm cells are highly heterogeneous based on their locations, and keep in mind the overlap between CD103+CD69+ Trm cells and activated CD103+ cells in human kidneys.
Potential function of tissue-resident memory T cells in inflammatory diseasesThe functional properties of Trm cells in diverse tissues have been widely explored74, 81-90, and it is likely that the heterogeneous subsets of CD8+ Trm cells contribute to persistent autoimmune disorders, such as in psoriasis, arthritis, cutaneous lupus erythematosus, autoimmune hepatitis and β-cell destruction in type 1 diabetes74, 84-87. It was shown that Trm cells produced cytokines, such as IFN-γ, that can stimulate B cells, natural killer cells and dendritic cells, and the ensuing innate immune cascade, therefore, added to the oxidative stress in different tissues81, 88. However, Trm cells also play a protection role in the regulation of immune homeostasis in non-barrier tissues (brain, liver) and barrier tissues (female reproductive tract and skin)81-83. In non-barrier tissues, such as the brain, CD8+ Trm cells have the ability to kill OVA-loaded targets82. Within the human liver, CD69+CD8+ effector memory T cells in the liver expressed a significantly higher level of the Trm-like phenotype (CD8+CD69+CD103+), with much lower cytotoxic proteins, as compared with peripheral effector cells83. This diminished cytolytic activity has been posited to protect the tissue (such as the brain and liver) from immune pathology when exposed to low or non-specific stimuli83, 91. Epithelial sites, including the FRT and skin, were intensively studied65, 81, 89. It was shown in an elegant study81 that lymphocytic choriomeningitis virus gp33 peptide-specific CD8+ Trm cells were established locally in the FRT of P14 chimeric mice, which have a transgenic T-cell receptor for lymphocytic choriomeningitis virus gp33-41 epitopes92. On challenge with recombinant vaccinia virus expressing OVA, a significantly reduction of viral load was detected within the FRT of mice harboring locally activated CD8+ Trm cells, as compared with naïve mice. Interestingly, memory T cells were found to be far higher on the mice skin that was pre-treated with a non-specific inflammatory stimulus65 and, thus, in the absence of antigen stimulation, inferring that Trm cells can be generated locally and provide protection without the existence of local antigen.
Knowledge of the functional features of Trm cells in kidney disease remains limited so far. An expansion of CD8+ Trm cells was observed in the kidneys of people with lupus nephritis or MRL/lpr mice (a model for an autoimmune disease resembling systemic lupus erythematosus), and correlated with kidney disease severity, providing the evidence that renal CD8+ Trm cells are involved in the pathogenesis of kidney injury93. Further studies are necessary to fill in the gap of showing discrete functional properties of Trm cells in inflammatory disease, and the beneficial effects might be able to be leveraged in specific tissue microenvironments of inflammatory disease, including DKD.
TREGSS IN DKDTregs make up 5–20% of the CD4+ T cells and include: (i) inducible Tregs produced in the periphery; and (ii) natural Tregs produced in the thymus94. It is reported that inducible Tregs implement their suppressive effect mainly by the effects of TGF-β1 and IL-1095. TGF-β1 can effectively induce the differentiation of Trm cells in the kidney96. As for IL-20, which is a pro-inflammatory cytokine of the IL-10 family, studies in diabetic mice showed increased IL-20 expression in renal podocytes, and anti-IL-20 mAb (7E) treatment reduced mesangial cell expansion and inflammatory responses, as indicated by a lower level of inducible nitric oxide synthase, tumor necrosis factor-α (TNF-α) and monocyte chemotactic protein-1 (MCP-1) expression in renal tissue97. Naturally arising Tregs express FOXP3 and among them, CD4+CD25+ Tregs are the most investigated subsets in the context of autoimmune diseases98, 99. CD4+CD25+FOXP3+Tregs maintain the self-tolerance immune balance and modulate a wide range of immune responses, including activation, proliferation and effector function98, 100. A recent study showed that adoptive transfer of FOXP3+ Tregs displayed an improved insulin sensitivity and a significant decline in albumin : creatinine ratio in the urine of db/db type 2 diabetic mice101, which implies that CD4+FOXP3+ Tregs might play a crucial part in reversing the progression of DKD. Furthermore, FOXP3+ Tregs can negatively regulate the effect of other T cells by competing for antigen-presenting cells through CTLA-4, whereby the administration of CTLA4-Fc (abatacept) subcutaneously further validated the protection effect of FOXP3+ Tregs, showing reduced levels of albuminuria in high-fat diet type 1 diabetic mice50. These findings suggest that the Tregs might represent a protective regulator in the development of DKD.
An appropriate ratio between pro-inflammatory and anti-inflammatory factors is critical to maintain a balanced microenvironment and reduce the risk of inflammatory disease. Th17 cells can produce IL-17, which has been shown to contribute to the pro-inflammatory progression of diabetes30. In contrast, Tregs cells are known to be potent suppressors of autoimmunity, and reported to dampen Th1, Th2 and Th17 cells response, specifically102-104. In people with type 2 diabetes and/or impaired kidney function, a reduced level of peripheral Tregs and an elevated serum Th17 : Tregs ratio were seen compared with controls, and positively related to the urine albumin : creatinine ratio104-107. Collectively, more studies are required to elucidate the clinical application of Tregs as a potential cellular immunotherapy for people with DKD, such as selective treatment of low-dose IL-2 to modulate CD4+ Tregs.
SIGNATURE T-CELL-RELATED CYTOKINES IN DKDIt has been shown that systematic and local inflammation are implicated in the development of DKD, and specific cytokines are widely studied22, 108-117. In reverse, in vitro and in vivo studies showed that short-term hyperglycemia can persistently exacerbate the inflammation response by activating epigenetic mark, histone 3 lysine 4 monomethylation (H3K4me1) in the promoter of the nuclear factor-κB subunit p65, which controls cytokine production from diverse immune cells113. This pro-inflammatory cascade persists even in a subsequent normoglycemia environment113. Therefore, it is of paramount importance to identify signature cytokines and prognostic indicators for the renal injury associated with DKD.
IL-17AThe IL-17 family is composed of six homodimeric cytokines (IL-17A–F), which are mainly produced by activated memory T cells118, 119. IL-17A, commonly referred to as IL-17, is largely produced by T-helper cells (Th17), and was originally considered to have pro-inflammatory properties in type 1 diabetes, probably due to the influence of the inducible nitric oxide release on β-cells120, 121. Several studies reported an increased number of IL-17-producing cells, and production of IL-17 in the serum, spleen and pancreas of non-obese diabetic (NOD) mice (a model of type 1 diabetes mellitus) as diabetes progressed121-124. Similarly, a significantly higher level of IL-17 was also shown in the serum of people with type 1 diabetes and type 2 diabetes125-127. IL-17 deficiency was shown to play an integral role in suppressing the development of diabetes, as well as protection on the kidney, characterized by reduced albuminuria, glomerular injury and hypertrophy, macrophage accumulation, and renal fibrosis in STZ-induced diabetic mice (a model of type 1 diabetes)128. Mycophenolate mofetil suppressed the proliferation of IL-17A+CD4+ T cells (Th17 cells) during the development of DKD in STZ-induced diabetic mice from 8 to 16 weeks, whereas the number of IFN-γ+CD4+ Th1 cells was only decreased at an early stage, highlighting the importance of the involvement of Th17 cells compared with Th1 cells129. This downregulation of Th17 cells by mycophenolate mofetil was also found to be associated with albuminuria reduction129. Another study in line with the pathogenesis impact of IL-17A found that structural lesions, including mesangial matrix accumulation and glomerular basement membrane thickening, can be ameliorated with neutralizing antibody against IL-17A30. Together, these findings suggest the critical regulatory role of IL-17A in immune diseases and the promising effect of IL-17A neutralization on DKD treatment. Nevertheless, many other studies have shown controversial results. A low dose of IL-17A protected STZ diabetic mice from kidney disease, and a more pronounced renal change, such as tubular injury, interstitial fibrosis and mesangial expansion, was observed in IL-17A knockout STZ diabetic mice compared with wild-type mice130. Furthermore, the transfer of adoptive IL-17-producing γδT cells in NOD mice showed that it did not aggravate diabetes, but protected NOD mice by decreasing its incidence of diabetes131. In human studies, there was no significant difference between the level of serum IL-17 in people with type 2 diabetes mellitus and those without diabetes105, 132. Additionally, in a cross-sectional study of Asian and Indian populations, the level of serum IL-17 was significantly lower in people with diabetes with or without renal lesions133. Regarding the possible mechanism, an elegant study showed that CD4+ Th17 cells were capable of co-secreting IL-10, playing regulatory properties, and stimulus with TGF-β and IL-6 can completely abrogate the pathogenic function of Th17 cells, despite a rise of IL-17 in vitro and in vivo134. Hence, it is potentially because IL-17-producing Th17 cells might produce other cytokines that play a greater role in determining the pathological or tolerogenic effects, or the role of IL-17 might be mitigated by other cytokines in different microenvironments of the disease course. Therefore, further evaluation is still required to consolidate the dose–effect of IL-17A and subsequent inflammatory signaling pathway in DKD.
IL-2The pathological role of IL-2/IL-2 receptor (IL-2R) in kidney injury has long been shown in early studies135. It was shown that serum soluble IL-2R (sIL-2R) levels increased in people with renal dysfunction, and a significant negative correlation was observed between sIL-2R and creatinine clearance136. In vitro activated T cells showed marked upregulation of binding affinity and surface receptor number under exposure to advanced glycation end-products (oxidative derivatives resulting from hyperglycemia), and an elevated expression of different pro-inflammatory Th1 cytokines, such as IFN-γ and IL-2135. In patients with DKD and overt nephropathy (daily urine protein loss >3.5 g/day), a significantly higher level of serum IL-2R was presented compared with those with normoalbuminuria (<30 mg/day) or microalbuminuria (30–300 mg/day)33. This evidence suggests that IL-2R acts in concert with other Th1 cytokines, and might be an important driver in the pathological development of kidney injury in DKD33.
Given the fact that IL-2 is essential for the development and normal function of Tregs, which are critical in preventing autoimmune diseases, it is feasible to make an effort to explore an efficient strategy of IL-2, or analogs of IL-2, treatment in people with type 1 diabetes. A recent study reported that low-dose mouse IL-2/CD25 (mIL-2/CD25) prevented diabetes in NOD mice and regulated diabetes in hyperglycemic mice137. However, studies of low-dose IL-2 in chronic kidney disease in people with type 2 diabetes are scant. Studies showed that low-dose IL-2 selectively expanded CD4+CD25+FOXP3+ Tregs in patients with chronic kidney disease, and these Tregs hampered the production of pro-inflammatory Th1 and Th17 cells104; hence, it will be interesting to explore whether low-dose IL-2 can provide beneficial effect to patients with DKD.
TNF and TNF receptorTNF was identified five decades ago138, 139 as the product of monocytes that induced acute and chronic systematic inflammatory responses140-142. Apart from hematopoietic cells, TNF has also been found in intrinsic renal parenchymal cells, glomerular visceral epithelial cells and mesangial cells143-145. The TNF superfamily and its receptors on the surface of T cells are of paramount significance to normal T-cell function, as discussed elsewhere146, 147.
TNF was first shown to have profound pro-inflammatory effects in DKD in 1991148. Results from that study demonstrated that the incubation of peritoneal macrophages from normal rats with glomerular basement membrane from the diabetic group showed the ability to synthesize increased levels of TNF and IL-1, compared with macrophages incubated with glomerular basement membrane from normal rats. This finding indicates that advanced glycation end-products, which were generated on the glomerular basement membrane in the setting of diabetes, might strongly boost the production of these cytokines, and could contribute to the alteration of glomerular microcirculation, as shown by substantially increased urinary albumin excretion in diabetic mice. Indeed, TNF-α microribonucleic acid and protein levels in the renal interstitial fluid and urine were augmented before a significant elevation of urinary albumin excretion in a STZ-induced diabetic rodent model149-151. A positive relationship between urinary levels of TNF-α and urinary albumin excretion was further confirmed, with a reciprocal role for albuminuria on the production of TNF-α150. Conversely, TNF antagonist was reported to reduce urinary excretion of TNF, likely by preventing Na retention (which was shown to promote hypertension and cause renal hypertrophy by TGF-β effects152, 153) in distal tubule cells through an increase of tubular Na transport and sodium-dependent solute uptake154, and attenuated the loss of kidney function in a diabetic rat model150, 155. Similarly, in clinical trials, many studies showed that the serum or urinary levels of TNF-α in people with type 2 diabetes mellitus were higher than those without diabetes156-158. These changes show the possibility that TNF-α might serve as a promising marker in predicting the risk for progressive kidney impairment in DKD.
There has been an increasing appreciation of the importance of TNF receptor (TNFR) as a predictor of the development and progression of DKD for both type 1 diabetes and type 2 diabetes159-162. TNFR type 1 and TNFR type 2 are the two main distinct receptors of TNF that existed in both a membrane-embedded pattern and a soluble form in serum163. A small pilot study showed that an increase in soluble TNFR type 1 level is independently associated with an early decline in the estimated glomerular filtration rate (eGFR)159. For advanced kidney disease, an 8- to 12-year follow-up study has shown that circulating TNFR type 1 and TNFR type 2 levels, but not free or total TNF-α levels, were strongly associated with the risk of progression to ESKD in type 2 diabetes patients160. This relationship was found to be independent of other known risk factors or markers for progression of DKD, such as albuminuria160. These results have challenged the value of TNF-α levels as a DKD-related biomarker. As such, further studies in animal models of diabetes are required to determine the factors that drive an increase in TNFR, as well as the significance and causal relationship of these changes in the pathogenesis of DKD.
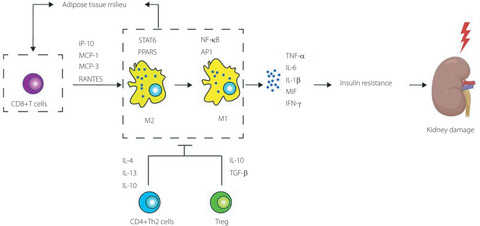
THERAPEUTIC IMPLICATIONS OF TARGETING CD8+ T CELLS IN ADIPOSE TISSUE IN DKDThe change of the microenvironment in diabetes brings about cellular alterations in adipocytes, and affects the cross-talk between adipose tissue and other organs, including the kidney, referred to as the adipo–renal axis164. Plenty of evidence has shown that obesity might be a risk factor for ESKD in people with type 2 diabetes and hypertension165-168. In people with DKD and obesity who had an eGFR <40 mL/min/1.73 m2 and urine albumin excretion <30 mg/day, a short-term intensive ketogenic diet improved their glomerular filtration rate, as well as metabolic markers, including fasting insulin, fasting glucose and insulin resistance, which shows that alteration in adipocytes might be a contri
留言 (0)