
記住我
 Huang, Zhang, Jiang, Hu, Sun, Zhang, and Ding: Inhibition of SKP2 Sensitizes Bromocriptine-Induced Apoptosis in Human Prolactinoma Cells
Inhibition of SKP2 Sensitizes Bromocriptine-Induced Apoptosis in Human Prolactinoma Cells
Abstract
Purpose
Huang, Zhang, Jiang, Hu, Sun, Zhang, and Ding: Inhibition of SKP2 Sensitizes Bromocriptine-Induced Apoptosis in Human Prolactinoma Cells
Inhibition of SKP2 Sensitizes Bromocriptine-Induced Apoptosis in Human Prolactinoma Cells
Abstract
Purpose
Prolactinoma (prolactin-secreting pituitary adenoma) is one of the most common estrogen-related functional pituitary tumors. As an agonist of the dopamine D2 receptor, bromocriptine is used widely to inhibit prolactinoma progression. On the other hand, it is not always effective in clinical application. Although a dopamine D2 receptor deficiency contributes to the impaired efficiency of bromocriptine therapy to some extent, it is unknown whether there some other underlying mechanisms leading to bromocriptine resistance in prolactinoma treatment. That is the main point addressed in this project.
Materials and MethodsHuman prolactinoma samples were used to analyze the S-phase kinase associated protein 2 (SKP2) expression level. Nutlin-3/adriamycin/cisplatin-treated GH3 and MMQ cells were used to analyze apoptosis in SKP2 overexpression or knockdown cells. SKP2 expression and the interaction partners of SKP2 were also detected after a bromocriptine treatment in 293T. Apoptosis was analyzed in C25 and bromocriptine-treated GH3 cells.
ResultsCompared to normal pituitary samples, most prolactinoma samples exhibit higher levels of SKP2 expression, which could inhibit apoptosis in a p53-dependent manner. In addition, the bromocriptine treatment prolonged the half-life of SKP2 and resulted in SKP2 overexpression to a greater extent, which in turn compromised its pro-apoptotic effect. As a result, the bromocriptine treatment combined with C25 (a SKP2 inhibitor) led to the maximal apoptosis of human prolactinoma cells.
ConclusionThese findings indicated that SKP2 inhibition sensitized the prolactinoma cells to bromocriptine and helped promote apoptosis. Moreover, a combined treatment of bromocriptine and C25 may contribute to the maximal apoptosis of human prolactinoma cells.
Introduction Pituitary prolactinoma, which is also known as prolactin-secreting pituitary adenoma, is one of the most common pituitary tumors that occurs in almost 0.4% of the general population [1]. As an estrogen-related functional tumor, pituitary prolactinoma is featured for hyperprolactinemia owing to the dysfunction of lactotrophic cells in the anterior pituitary gland [2]. In addition, gene mutations in lactotrophic cells were reported to play a critical role in prolactinoma tumorigenesis [3], which is accompanied by dysregulation and over-activation in multiple signaling pathways, such as Ras/mitogen-activated protein kinase, phosphoinositide 3-kinase, and transforming growth factor α cascades. The abnormities of these pathways further contribute to prolactinoma formation and pathological hyperprolactinemia [3]. In addition, estrogen hypersecretion can induce pituitary hyperplasia, lactotrophic prolactin (PRL) oversynthesis, and even estrogen-related tumors by binding to the estrogen receptors [4,5]. As estrogen receptor expression in prolactinoma is much higher than that of other pituitary tumor types [6], the most efficient way for prolactinoma therapy at present is dopamine agonists, in which bromocriptine is used most widely. In principle, bromocriptine binds to the dopamine D2 receptor on pituitary epithelial cells to inhibit PRL secretion and tumor cell growth [7,8]. On the other hand, although bromocriptine has been proven to inhibit prolactinoma progression by blocking the dopamine D2 receptor, it is not always effective in clinical applications. Bromocriptine resistance has been observed in 5%-18% patients with prolactinoma [9,10]. Although it has been reported that a dopamine D2 receptor deficiency or the impaired binding to the D2 receptor contributes to bromocriptine resistance [11], the precise molecular mechanism about bromocriptine resistance in prolactinoma is not completely understood. Recently, it was reported that the lower efficiency of bromocriptine therapy might be correlated with the abnormal expression of some proteins, such as PRDM2 and ESM1 [12,13]. Therefore, this study examined whether some specific overexpressed oncogenes compromise the proapoptosis effect of bromocriptine in prolactinoma. From this aspect, new strategies are needed to sensitize bromocriptine in promoting the apoptosis of PRL-secreting pituitary cells. Apoptosis induction is one of the most important strategies for tumor therapy. Among the apoptosis-inducing factors, p53 is the most famous. Previous studies have shown that p53 transactivates a broad range of proapoptotic proteins to promote cell apoptosis, such as Bax, Bid, Puma, 14-3-3σ, and p21 [14,15]. Although p53 expression is reported to be associated with the tumor invasiveness in pituitary tumors [16], there are few reports on the role of p53-dependent apoptosis in pituitary tumor therapy [17]. As a proto-oncoprotein, S-phase kinase associated protein 2 (SKP2) is overexpressed in a variety of human cancers, including lymphomas, prostate cancer, colorectal cancer, melanoma, pancreatic cancer, and breast carcinoma [18]. On the other hand, the role of SKP2 in pituitary tumors, especially prolactinoma, is not completely understood. SKP2 is the key component of the SKP1–Cullin1–F-box (SCF) E3 ligase complex [19], which is crucial for cell cycle progression by targeting p27. Furthermore, several other substrates of SKP2 have been verified, such as p21, p57, and c-Myc, most of which are tumor suppressor proteins [18]. Note that the anaphase-promoting complex (APC), a pivotal E3 ligase that is activated by interacting with Cdh1, functions in degrading SKP2 [20]. Therefore, the protein stability of SKP2 remains when it interacts with cullin 1 (Cul1) and forms the SKP2 containing SCF (SKP1–Cul1–F-boxSKP2) complex. Alternatively, SKP2 can be degraded in the APCCdh1 complex. In addition, accumulating evidence shows that SKP2 is involved in regulating cellular apoptosis in a range of human cancer cells by suppressing the p53 or p27 pathway [21]. Based on these findings, this study examined whether SKP2 could participate in prolactinoma cell apoptosis and act as a candidate target gene for prolactinoma therapy.In this study, two well-known PRL-secreting rat pituitary cell lines, GH3 and MMQ, were used to study the probable mechanism that coordinates SKP2 expression and bromocriptine therapy in prolactinoma. This study first identified that prolactinoma samples have high protein levels of SKP2, which inhibit GH3 and MMQ cell apoptosis in a p53-dependent manner. In addition, bromocriptine treatment prolonged the half-life of SKP2, which resulted in SKP2 overexpression to a greater extent. As the overexpression of SKP2 compromised bromocriptine-induced apoptosis, a combination of bromocriptine treatment and SKP2 inhibitor C25 induces the maximal apoptosis. These results indicate that Skp2 knockdown sensitized bromocriptine in promoting the apoptosis of PRL-secreting pituitary cells.
Materials and Methods 1. Pituitary tumors samples All the pituitary adenoma samples were obtained from the Department of Neurosurgery, Shanghai Institute of Neurosurgery, Changzheng Hospital, Second Military Medical University (Shanghai, China). Pituitary tumors (n=81) removed at transsphenoidal surgery were classified histologically using hematoxylin and eosin and immunohistochemical hormone staining. Benign adenomas were categorized as growth hormone–secreting tumors (n=19), adreno-corticotropic-hormone–secreting tumors (n=13), prolactinomas (n=23), and non-functioning pituitary adenomas (NFPAs; n=26). The pituitaries removed at autopsy were verified by the Forensic Authentication Center of Changzheng Hospital and used as normal controls (n=4). Table 1 lists the patients and sample characteristics. The definition of high or low SKP2 expression in Table 2 was based on immunohistochemistry staining, and representative staining pictures are shown in Supplementary Fig. S1. Scores 0 and 1 were defined as low SKP2 expression; scores of 2 and 3 were defined as high SKP2 expression. 2. Material and reagentsBromocriptine mesylate was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Nutlin-3, doxycycline, propidium iodide, cycloheximide, adriamycin, and cisplatin were obtained from Sigma-Aldrich (St. Louis, MO). G418 sulfate was supplied by Life Technologies (Grand Island, NY). C25 was purchased from Xcess Biosciences Inc. (San Diego, CA). The luciferase activity was determined using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI). The Skp2 siRNA for rat Skp2, p53 shRNA for rat p53, and caspase 2 siRNA for rat caspase 2 were supplied by Santa Cruz Biotechnology.
3. Plasmids3xflag-Skp2, Myc-Cul1, Myc-CDH1, and HA-Ubiquitin were cloned from the rat cDNA library and verified by sequencing. A reporter containing the p53-binding site placed upstream of the firefly luciferase gene (PG13-Luc) and reporters containing a mutation on p53-binding site (MG15-Luc) were acquired from Addgene (Cambridge, MA).
4. SKP2 inducible systemThe doxycycline-inducible SKP2 expression lentiviral systems were constructed according to the instructions of Lentiviral Tet-On 3G Inducible Expression Systems (Clontech, Mountain View, CA). The GH3 cells were infected with the above virus and a stable Tet-On inducible cell line was selected by G418 addition according to the manufacturers’ instructions.
5. Immunoblot analysisThe cell and tissue protein were prepared with 1% NP-40 cell lysis buffer (50 mM Tris HCl [pH 8.0], 120 mM NaCl, 1% NP-40) containing 1 mM dithiothreitol, and phosphatase inhibitor cocktails I and II (Sigma). The protein was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis for further analysis. The following lists the primary antibody information: anti-SKP2 (H435, Santa Cruz Biotechnology), anti-p53 (FL-393, Santa Cruz Biotechnology), anti-cleaved caspase 3 (ab2302, Abcam, Cambridge, MA), anticytochrome C (ab13575, Abcam), anti-Bax (N-20, Santa Cruz Biotechnology), anti–cleaved poly(ADP-ribose) polymerase (PARP; 9545, Cell Signaling Technology, Beverly, MA), antiactin (I-19, Santa Cruz Biotechnology), anti-Flag (F7425, Sigma-Aldrich), anti–c-Myc (9E10, Santa Cruz Biotechnology), anti-p21 (ab18209, Abcam), anti-Puma (ab9643, Abcam), and anti-Mdm2 (SMP14, Santa Cruz Biotechnology). The quantified relative ratios of remaining SKP2 were obtained by densitometry using Image J software, and the data at 0 hour was set to 1.
6. Generation of stable p53 knockdown GH3 cell lineThe synthetic sequences targeting either the rat p53 gene were inserted into pLVX-IRES-Neo plasmid (Clontech). A lentivirus was then generated using lentiviral packaging systems (Clontech) and the GH3 cells were infected with the p53 knockdown lentivirus. A stable cell line was generated and cultured by the addition of G418 to select a positive stable knockdown clone. The core sequences for targeting p53 was 5′-GGTGGAAGGAAATCCGTAT-3′. The expression of p53 in stable knockdown cell lines was confirmed by immunoblotting and real-time polymerase chain reaction (PCR).
7. Co-immunoprecipitation assayAll the co-immunoprecipitation steps were performed at 4°C. The cells were washed three times and then lysed in 1% NP-40 cell lysis buffer for 30 minutes. The cell extracts were centrifuged to obtain the supernatant. The supernatant was then added with the indicated antibody overnight, and then incubated with Protein A/G Plus Agarose (Santa Cruz Biotechnology) for 5 hours. The immunoprecipitate was washed three times in cell lysis buffer, followed by immunoblotting analysis.
8. Cell culture and transfectionThe rat GH3 and MMQ pituitary adenoma cell line was obtained from the American Type Cell Collection. The GH3 and MMQ cells were propagated in the F12K medium (Invitrogen, Carlsbad, CA) supplemented with 2.5% fetal bovine serum, 15% horse serum, 100 units/mL penicillin, and 100 units/mL streptomycin. All cell lines were cultured at 37°C in a humidified atmosphere containing 5% CO2. The cells were transfected with the control or Skp2 siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Forty-eight hours after transfection, the cells were collected and the transfection efficiency was determined by quantitative real-time PCR.
9. Analysis of cell apoptosis and cell numberThe caspase 3/7 activity was determined using a Caspase-Glo 3/7 Assay Kit (Promega) according to the instructions supplied, the final relative caspase 3/7 activity was normalized to the viable cell number determined using a CellTiter-Fluor Cell Viability Assay kit (Promega). Cell apoptosis was detected using an Annexin V-FITC Apoptosis Detection Kit (Sigma-Aldrich) according to the manufacturer’s instructions and determined by fluorescence-activated cell sorting. For cell number analysis, 1×103 GH3 cells were seeded in each well of a 96-well plate and the number of viable cells were assessed using a CellTiter-Fluor Cell Viability Assay Kit (Promega).
10. Quantitative real-time PCRThe total cellular RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s protocol, and the quantity and quality were measured by spectrophotometry. The reverse transcription (RT) reactions were performed with 1 μg of RNA using the RT reagents (Takara, Dalian, China) to synthesize cDNA according to the manufacturer’s instructions. Quantitative PCR was performed according to the manufacturer’s instructions using a SYBR green mixture (TOYOBO, Osaka, Japan).
11. Statistics Each experiment was performed independently in triplicate. All the data are expressed as the means±standard deviation. The statistical significance of the differences between the groups was analyzed by a Student’s t test using SPSS ver. 10.0 (SPSS Inc., Chicago, IL), except in Table 2 where a Pearson’s chi-square test was used. Results 1. SKP2 was overexpressed in pituitary prolactinoma SKP2 is overexpressed in many human cancers, in which SKP2 promotes cancer progression and acts as an oncoprotein. As a result, this study examined whether SKP2 also plays a critical role in pituitary adenomas. SKP2 might be overexpressed in pituitary prolactinoma samples, which is reported in two open Gene Expression Omnibus (GEO) datasets. To verify these findings, SKP2 gene expression was detected in 81 pituitary tumor samples collected from transsphenoidal surgery (Tables 1 and 2) and four normal pituitary samples collected from autopsy. All the pituitary tumor samples were divided into four groups—growth hormone secreting tumors, adreno-cortico-tropic-hormone secreting tumors, PRL secreting tumors (prolactinomas), and NFPA by a histologic examination and hormone staining. Surprisingly, the SKP2 gene was greatly up-regulated in prolactinoma, with a mean ~10 times higher than that of the normal control pituitary samples (p Fig. 1A). Furthermore, the Skp2 mRNA level was up-regulated moderately in NFPA. In contrast, there were no significant Skp2 mRNA changes in the GH and adreno-cortico-tropic-hormone pituitary tumors compared to their respective normal pituitary controls. (Fig. 1A). As the SKP2 gene was enhanced dramatically in PRL at the mRNA level, SKP2 protein expression was next examined in the same samples. To this end, the samples with the top two levels of SKP2 expression at the mRNA level in Fig. 1A were chosen to analyze SKP2 protein expression in each group. Consistently, expression of the SKP2 protein was much higher in PRL than that in any other groups (Fig. 1B and C). In addition, the relationship between the clinicopathological features and SKP2 expression was analyzed and a clear correlation between SKP2 expression and prolactinoma tumor size was observed (Table 2). Collectively, these results suggest that both mRNA and protein expression of SKP2 were overexpressed greatly in pituitary prolactinoma. Although SKP2 was also overexpressed in NFPA, this study focused on PRL, in which the SKP2 expression level was up-regulated the highest among the different kinds of pituitary tumors. 2. Overexpression of SKP2 suppresses p53-mediated apoptosis in PRL-secreting pituitary cell lines SKP2 was reported to suppress apoptosis in many cancer cells [22]. This prompted the present study to test whether SKP2 is also involved in apoptosis regulation in pituitary cell lines, particularly in PRL-secreting pituitary cell lines. First, a stable Tet-On inducible GH3 cell line, a rat PRL secreting cell line, in which SKP2 was overexpressed by adding doxycycline into the culture medium, was generated (Fig. 2A). As SKP2 was reported to suppress apoptosis through the p53 pathway [21], the changes in the p53 target genes in SKP2 inducible GH3 cells were next examined. Interestingly, SKP2 overexpression in GH3 cells suppressed the protein expression of the p53 target genes, including Puma, Bax, Mdm2, and p21 (Fig. 2B, lanes 1 and 3). Consistent with previous reports, the expression of p53 and its target genes in GH3 cells was up-regulated by a nutlin-3 treatment, a compound that activates p53 selectively by stabilizing its protein expression [21]. On the other hand, SKP2 overexpression significantly suppressed the protein expression of the p53 target genes (Fig. 2B, lanes 2 and 4) under nutlin-3 stimulation. In addition, the mRNA levels of the p53 target genes, such as Bax, Puma, 14-3-4σ, p21, and Mdm2 were all down-regulated by SKP2 overexpression regardless of the presence of nutlin-3 stimulation (Fig. 2C). In GH3 cells, the induced SKP2 overexpression suppressed the activity of a reporter containing p53-response elements both with and without nutlin-3 stimulation, but this suppressive effect was lost in the cells transfected with a reporter containing the mutant p53-response elements (Fig. 2D). These results suggest that SKP2 suppresses the transactivation of p53. As p53 is the central node of apoptosis regulation [23], this study tested whether SKP2 overexpression affects apoptosis in GH3 cells. The activities of caspase 3/7, the key markers of apoptosis [24], were attenuated significantly in the SKP2-overexpressed GH3 cells both with or without the nutlin-3 treated treatment (Fig. 2E). Furthermore, the DNA damaging agent, adriamycin, could also increase the expression of p53, Puma, and proapoptosis protein BAX, cleaved caspase 3. This enhancement, however, was abrogated by the overexpression of SKP2 (Fig. 3A). The overexpression of SKP2 also inhibits Puma and p21 mRNA expression with or without adriamycin stimulation (Fig. 3B). Similarly, either the adriamycin- or cisplatin-induced caspase 3/7 cleavage was also inhibited significantly by SKP2 overexpression (Fig. 3C). Overall, the overexpression of SKP2 suppresses PRL-secreting pituitary cell apoptosis in a p53-dependent manner. 3. Skp2 knockdown synergizes with the DNA damage induction agents to promote apoptosis in PRL-secreting pituitary cells Next, this study examined whether the knockdown of Skp2 promotes apoptosis in GH3 cells. Consistent with this hypothesis, a significant increase in caspase 3/7 activity was observed in the Skp2 knockdown GH3 cells (Fig. 3D and E). Furthermore, the knockdown of Skp2 also increased the caspase 3/7 activity in MMQ cells, which is another rat pituitary cell line secreting only PRL (Fig. 3E). These results suggest that Skp2 knockdown promotes pituitary cell apoptosis. DNA damage–induced cancer cell apoptosis is one of the most efficient ways of cancer therapy. Therefore, this study examined whether the knockdown of Skp2 and the induction of DNA damage contributes to cell apoptosis synergistically. As shown in Fig. 3E, increased caspase 3/7 activity was observed when the pituitary cells were treated with adriamycin or cisplatin alone. Interestingly, the knockdown of Skp2 could robustly enhance the adriamycin or cisplatininduced caspase 3/7 activity (Fig. 3E). In addition, a stable p53 knockdown GH3 cell line (GH3/p53kd), in which only ~10% of p53 expression was observed compared to the wild-type GH3 cells, was constructed to determine if the promotion of DNA damage–induced apoptosis by Skp2 knockdown is mediated by the p53 pathway (data not shown). Surprisingly, the knockdown of Skp2 specifically enhanced the adriamycin-induced caspase 3/7 activity only in the wild-type GH3 cells, but not in the GH3/p53kd cells (Fig. 3D and F), indicating that Skp2 knockdown promotes DNA damage– induced apoptosis in a p53 dependent manner. Collectively, these results indicate that the knockdown of Skp2 synergizes with the DNA damage induction agents to promote apoptosis in PRL-secreting pituitary cells. 4. Bromocriptine promotes SKP2 expression by inhibitingubiquitination and degradation As one of the most widely used dopamine agonists in prolactinoma therapy, bromocriptine can effectively induce apoptosis in pituitary cells [25]. Based on the results that the knockdown of Skp2 is beneficial to apoptosis, this study next examined whether bromocriptine mediated pituitary cell apoptosis is caused by downregulating SKP2. Surprisingly, upon the bromocriptine treatment, the SKP2 protein level in GH3 cells was up-regulated robustly (Fig. 4A). On the other hand, no changes in Skp2 mRNA expression were observed (Fig. 4B). Therefore, this study examined whether bromocriptine affects SKP2 degradation in pituitary cells. Consistent with this hypothesis, the bromocriptine treatment prolonged the SKP2 half-life significantly (Fig. 4C). The underlying mechanism through which bromocriptine inhibited SKP2 degradation was next examined. Previous studies showed that SKP2 is degraded mainly by the APC, which is a pivotal E3 ligase that is activated by binding with Cdh1 [20]. In contrast, the protein stability of SKP2 remains when it interacts with Cul1 and forms the SKP1–Cul1–F-boxSKP2 complex, which is essential for the ubiquitination of a broad range of proteins [26]. The apparently opposite destiny of SKP2 prompted an examination determine if the interaction between SKP2 and Cul1 or Cdh1 was affected by the bromocriptine treatment. In Fig. 4D, the interaction between SKP2 and Cul1 was increased dramatically by bromocriptine in a dose dependent manner by MG132 addition. In contrast, bromocriptine abrogated the interaction between SKP2 and Cdh1 in a dose dependent manner (Fig. 4E). These results suggest that the bromocriptine treatment promotes SKP2 and Cul1 interaction and suppresses the interaction between SKP2 and Cdh1. In other words, bromocriptine reduces the degradation of SKP2 through its effect on the interaction balance between SKP2 with Cul1 or Cdh1. Consistent with these results, bromocriptine inhibited SKP2 ubiquitination robustly (Fig. 4F). These results support the hypothesis that bromocriptine inhibits the ubiquitination and degradation of SKP2 in pituitary cells. 5. Inhibition of SKP2 sensitize bromocriptine-induced apoptosis in GH3 cells Based on previous results, the bromocriptine treatment enhanced the protein expression of SKP2, which might in turn impair its pro-apoptosis effect. Therefore, this study examined whether a combination of a bromocriptine treatment with Skp2 knockdown could induce the maximum apoptosis in prolactinoma cells. Chan et al. [27] reported that C25, a small compound, could specifically inhibit the SKP2 activity, but not other SCF complexes. Consistent with this report, the expression of SKP2 in GH3 cells was inhibited significantly by the C25 treatment (Fig. 5A). Either bromocriptine or the C25 treatment alone could enhance the apoptosis activity in GH3 cells, as revealed by the increased expression of apoptosis markers, such as cleaved caspase 3, cytochrome C, Bax, and cleaved PARP (Fig. 5A, lanes 2 and 3). Surprisingly, the expression of these pro-apoptosis proteins was increased to the greatest extent when bromocriptine was combined with a C25 treatment (Fig. 5A, lane 4). In addition, C25-induced GH3 cell apoptosis could be enhanced significantly by a combined treatment of bromocriptine in a dose-dependent manner (Fig. 5B). In contrast, the number of viable cells was the lowest when GH3 cells were treated with C25 and the maximal dose of bromocriptine simultaneously (Fig. 5C). These results suggest that SKP2 overexpression in prolactinoma samples might be a potential factor that compromises the pro-apoptotic effects of bromocriptine on prolactinoma therapy. The doxycycline-inducible SKP2 expressing GH3 cells were used to test the hypothesis. It was found that bromocriptine increased the expression of multiple apoptosis markers, such as cleaved caspase 3, cytochrome C, Bax, and cleaved PARP (Fig. 5D, lanes 1 and 2). In contrast, the apoptosis activity in GH3 cells was inhibited dramatically by the overexpression of SKP2 regardless of the bromocriptine treatment (Fig. 5D, lanes 3 and 4). Furthermore, the percentage of apoptotic cells was decreased significantly by SKP2 overexpression regardless of the bromocriptine treatment (Fig. 5E). Consistently, the decline of the bromocriptine-induced cell number could be rescued markedly by SKP2 overexpression (Fig. 5F). In summary, bromocriptine-induced apoptosis in GH3 cells could be enhanced further by the inhibition of SKP2. In contrast, the overexpression of SKP2 compromised the bromocriptine-induced apoptosis in GH3 cells. DiscussionThis study first identified that SKP2 is overexpressed in prolactinoma subtype samples. Moreover, overexpressed SKP2 inhibits apoptosis in PRL-secreting cells through the p53 pathway. Second, the relationship between bromocriptine therapy and SKP2 in PRL-secreting pituitary cells was examined. As a bromocriptine treatment prolonged the halflife of SKP2 and resulted in SKP2 overexpression at the same time, the pro-apoptosis effect of bromocriptine was compromised to a certain extent. Third, a combination of the SKP2 inhibitor, C25, and bromocriptine resulted in the maximal apoptosis in GH3 cells, indicating SKP2 inhibition synergizes with bromocriptine in promoting the apoptosis of PRL-secreting cells.
SKP2 is overexpressed in many cancer cells [18]. In this study, both the mRNA and protein levels of SKP2 are overexpressed in pituitary adenoma samples, particularly in prolactinoma, with more than 10-fold up-regulation. In addition, SKP2 regulates apoptosis in PRL-secreting pituitary cells in a p53-dependent manner. Therefore, these results indicate that SKP2 might be a powerful target for prolactinoma therapy in the future. Therefore, this study examined the potential mechanism through which the SKP2 protein level was modulated by the bromocriptine treatment. Interestingly, the bromocriptine treatment ameliorated the protein degradation of SKP2 significantly. The SKP2 protein was reported to be stable in the SCFSKP2 complex. In contrast, it was degraded in the APCCdh1 complex [26]. Therefore, this study examined whether the interaction “switch” of SKP2 between APCCdh1 and SCFSKP2 is regulated by the bromocriptine treatment. Consistent with this hypothesis, after the bromocriptine treatment, SKP2 interacted with Cul1 but not Cdh1, so that the protein stability of SKP2 remained in GH3 cells. As a dopamine agonist, bromocriptine was reported to inhibit PRL secretion to shrink the tumor volume and induce apoptosis in GH3 cells [28]. On the other hand, SKP2 overexpression compromised the bromocriptine-induced apoptosis in prolactinoma cells. In other words, high levels of SKP2 expression in prolactinoma may be a challenge for effective bromocriptine therapy. Based on these results, a more efficient way was developed to induce PRL-secreting cell apoptosis, which is a combination of C25-induced SKP2 inhibition and bromocriptine treatment. This combination significantly induced cell apoptosis and decreased the number of cancer cells. Overall, this research not only provides direct evidence that SKP2 inhibition sensitizes the brobromocriptine-induced apoptosis in pituitary cells (Fig. 6), but also provides new view sights into bromocriptine therapy for prolactinoma. ConclusionSKP2 overexpression compromised the bromocriptine-induced apoptosis in prolactinoma cells. In contrast, SKP2 inhibition sensitized the prolactinoma cells to bromocriptine and helped promote apoptosis. These data, therefore, suggested that a combined treatment of bromocriptine and SKP2 inhibitor C25 might be an effective therapy for human pituitary prolactinoma.
Electronic Supplementary Material Supplementary materials are available at Cancer Research and Treatment website (http://www.e-crt.org). Conflicts of InterestConflict of interest relevant to this article was not reported.
AcknowledgmentsThis study was funded by a research grant from National Natural Science Foundation of China (No. 81372716).
Fig. 1. SKP2 was up-regulated in pituitary prolactinoma. (A) Real-time RT-PCR analysis for SKP2 expression in normal and different types of adenomatous pituitary tissues as indicated. (B) Representative western blotting showing protein expression of the SKP2 in pituitary adenoma samples. Samples with the two highest SKP2 gene expressions in Fig. 1A of each group were selected for protein analysis. (C) Immunohistochemistry of PRL and normal pituitary adenoma samples to detect the level of SKP2. SKP2, S-phase kinase associated protein 2; RT-PCR, reverse transcription polymerase chain reaction; PRL, prolactin; GH, growth hormone; ACTH, adreno-cortico-tropic-hormone; NFPA, non-functioning pituitary adenoma.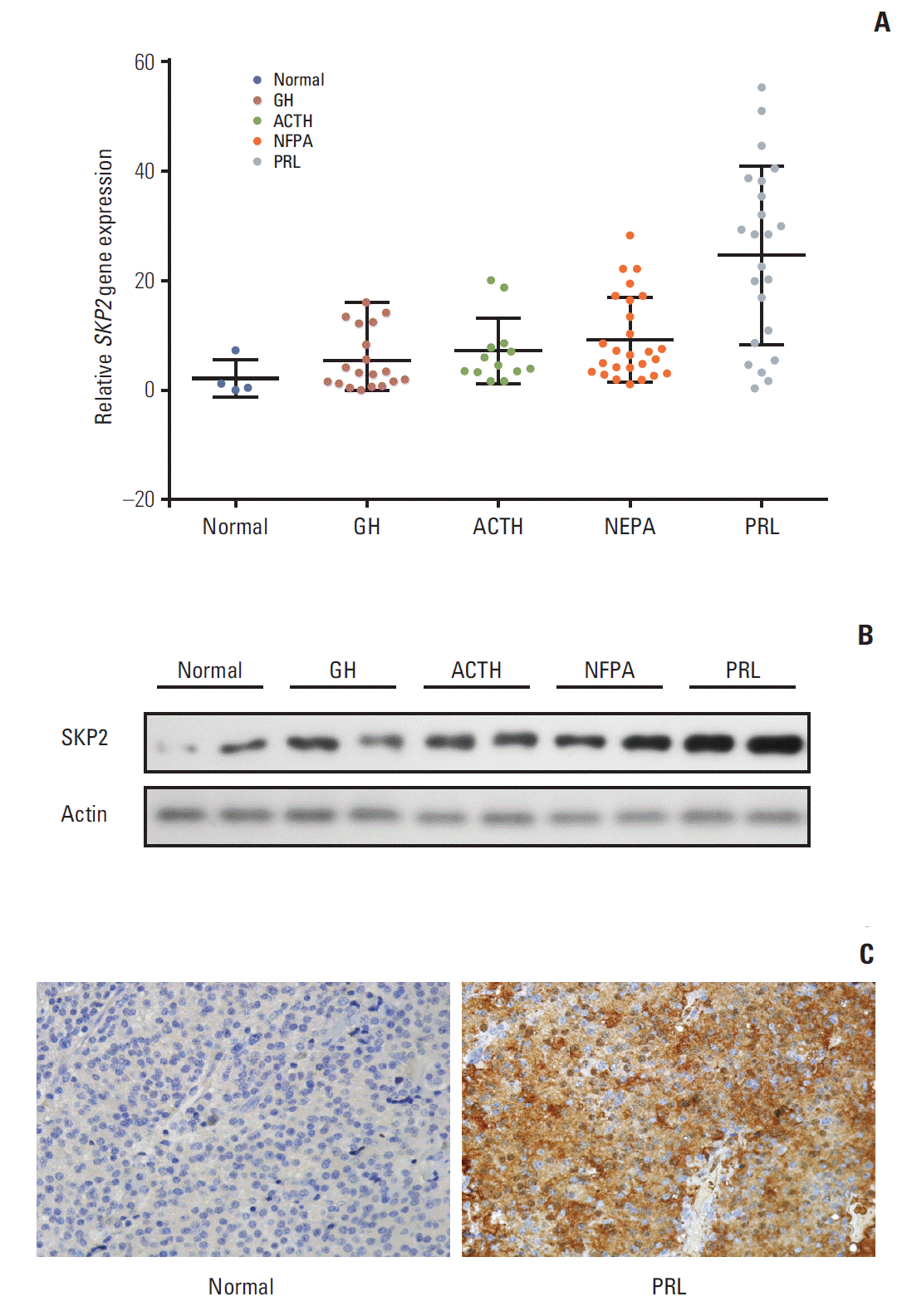 Fig. 2.
Fig. 2.
SKP2 overexpression suppresses apoptosis through p53 in GH3 cells. (A) GH3 cells infected with a lentiviral vector stably expressing the rat SKP2-inducible system. SKP2 was induced by 1 μg/mL doxycycline for 24 hours, and cells were then lysed and collected for immunoblotting analysis. (B, C). Twenty-four hours after inducing SKP2, the cells were treated with 5 μM nutlin-3 for 48 hours, and then lysed and collected either for immunoblotting analysis using the antibodies as indicated (B) or for quantitative real-time PCR analysis (C). (D) Twenty-four hours after inducing SKP2 expression, a reporter containing a synthetic p53-binding site (p53-luc) or reporter containing a mutation on the p53-binding site (p53MUT-luc) was transfected into the GH3 cells, together with a control reporter containing renilla luciferase. Twelve hours after transfection, the cells were treated with different concentrations of nutlin-3, as indicated for 48 hours, and the cells were then lysed and collected for luciferase activity analysis. (E) Twenty-four hours after SKP2 induction, the cells were treated with 10 μM nutlin-3 for 48 hours; apoptosis was determined and is shown by measuring the relative caspase 3/7 activity. SKP2, S-phase kinase associated protein 2; PCR, polymerase chain reaction; Dox, doxycycline. *p < 0.05, **p < 0.01, ***p < 0.001; ns, no significance.
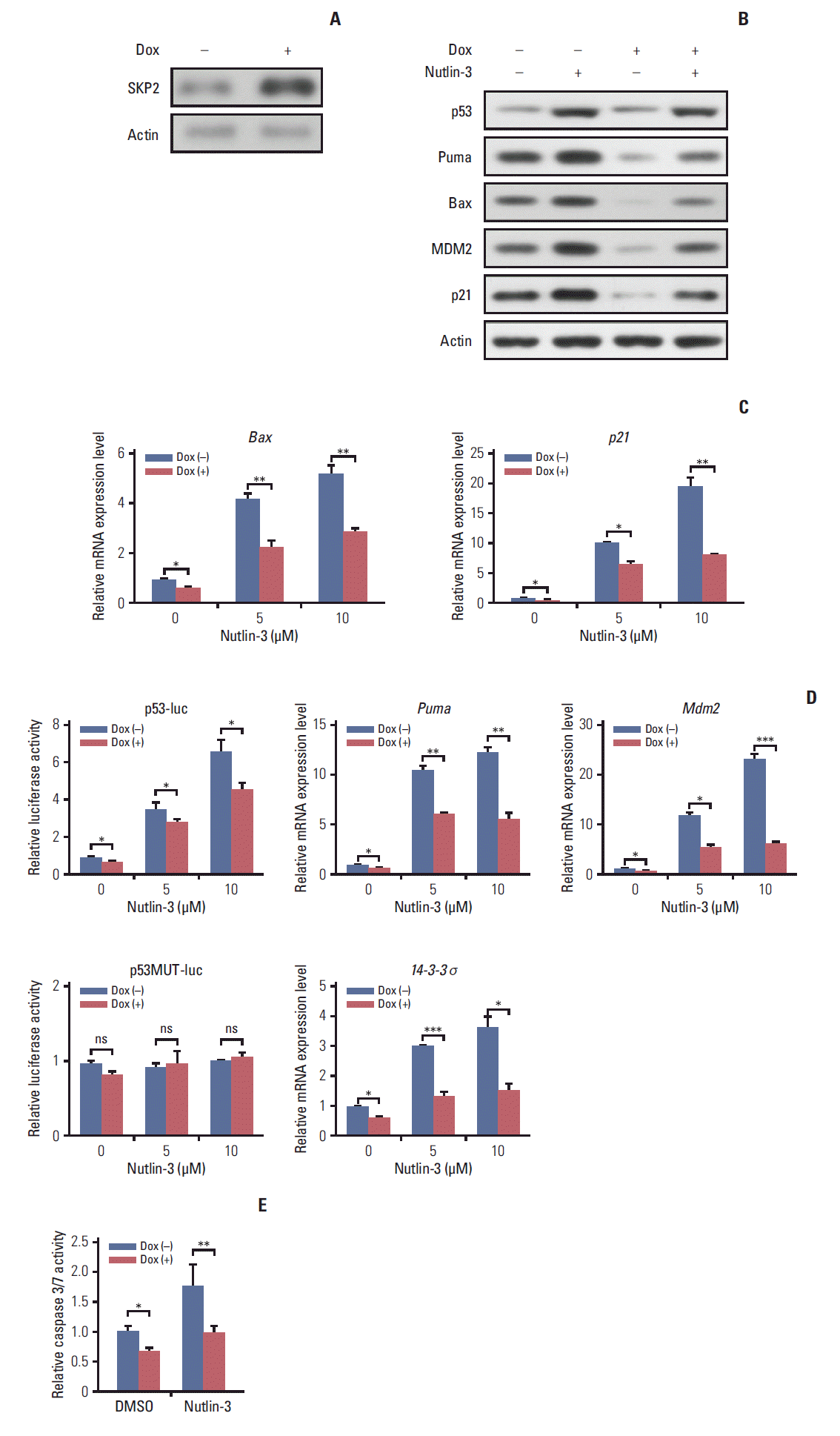 Fig. 3.
Fig. 3.
Skp2 knockdown synergizes with DNA damage induction agents to promote apoptosis in PRL-secreting pituitary cells. (A, B) Twenty-four hours after doxycycline addition, the GH3 cells were treated with 1 μg/mL adriamycin for 48 hours followed by either immunoblotting analysis using the antibodies as indicated (A) or real-time PCR analysis (B). (C) Twenty-four hours after SKP2 induction, the cells were treated with 1 μg/mL adriamycin or 5 μg/mL cisplatin for 48 hours in GH3 cells, followed by relative caspase 3/7 activity determination. (D) A control or Skp2 siRNA was transfected into the wild-type GH3 cells and stable p53 knockdown GH3 cells. Forty-eight hours after transfection, the cells were lysed and collected to analyze SKP2 expression by immunoblotting. (E, F) Twenty-four hours after transfection of the control or Skp2 siRNA, the cells were treated with 1 μg/mL adriamycin or 5 μg/mL cisplatin for 48 hours in the GH3 cells or MMQ cells (E), or indicated adriamycin concentration in wild-type GH3 cells and stable p53 knockdown GH3 cells (F), followed by a determination of the relative caspase 3/7 activity. SKP2, S-phase kinase associated protein 2; PRL, prolactin; PCR, polymerase chain reaction; Dox, doxycycline; DMSO, dimethyl sulfoxide; si-CTR, control siRNA. *p < 0.05, **p < 0.01, ***p < 0.001; ns, no significance.
 Fig. 4.
Fig. 4.
Bromocriptine stabilizes SKP2 expression by inhibiting SKP2 ubiquitination and degradation. (A, B) SKP2 expression in GH3 cells treated with or without 20 μM bromocriptine for 24 hours, the cells were collected and subjected to immunoblotting (A) or real-time PCR analysis (B). (C) The GH3 cells were transfected with Flag-SKP2. Twenty-four hours after transfection, the cells were treated with 10 μg/mL CHX and 20 μM bromocriptine for the indicated times. The cells were then lysed and subjected to immunoblotting analysis. The SKP2 levels from three independent assays were quantified by densitometry and the measurements were normalized to the start point (0 hour) to calculate the percentage changes following stimulation (C, lower panel). (D, E) the GH3 cells were transfected with Flag-SKP2 and Myc-Cul1 (D) or Flag-SKP2 and Myc-Cdh1 (E). Twenty-four hours after transfection, the cells were treated with 10 μM MG-132 and different concentration of bromocriptine, as indicated for 24 hours, and then cells were subjected to co-immunoprecipitation analysis. (F) The GH3 cells were transfected with Flag-SKP2 and HA-Ub. Twenty-four hours after transfection, the cells were treated with 10 μM MG-132 and 20 μM bromocriptine for 24 hours, and cells were then subjected to co-immunoprecipitation analysis. SKP2, S-phase kinase associated protein 2; CTR, control; PCR, polymerase chain reaction; CHX, cycloheximide; DMSO, dimethyl sulfoxide; HA-Ub, HA-ubiquitin. **p < 0.01, ***p < 0.001; ns, no significance.
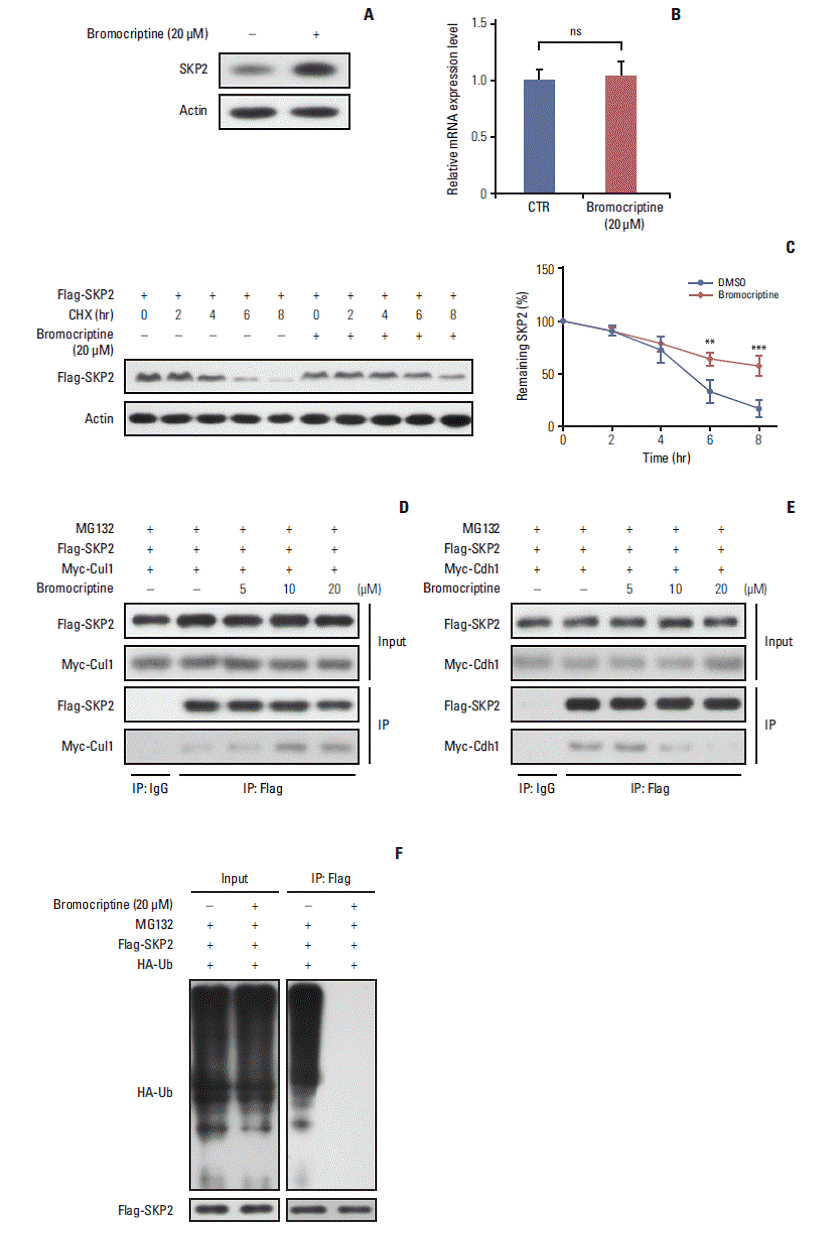 Fig. 5.
Fig. 5.
SKP2 inhibition sensitizes bromocriptine-induced apoptosis in GH3 cells. (A) GH3 cells were treated with 20 μM bromocriptine or 20 μM C25, as indicated for 48 hours. The cells were then lysed and collected for immunoblotting analysis using the indicated antibodies. (B) GH3 cells were treated with 20 μM C25 and different concentrations of bromocriptine, as indicated, for 48 hours. The cells were then lysed and collected for apoptosis determination by annexin-V staining and flow cytometry. (C) The GH3 cells were treated as the same in panel B, and cells were then subjected to determine the number of viable cells. (D) Twenty-four hours after SKP2 induction, the GH3 cells were subjected to 20 μM bromocriptine for 48 hours. The cell lysates were used for immunoblotting analysis using the indicated antibodies. (E) Twenty-four hours after SKP2 induction, the GH3 cells were exposed to the indicated bromocriptine concentrations for a further 48 hours, and cells were then lysed and collected for apoptosis determination by annexin-V staining and flow cytometry. (F) GH3 cells were treated as the same in panel F, and then subjected to a cell viability assay kit to determine the number of viable cells. SKP2, S-phase kinase associated protein 2; Cyt C, cytochrome C; PARP, poly(ADP-ribose) polymerase; DMSO, dimethyl sulfoxide; Dox, doxycycline. *p < 0.05, **p < 0.01, ***p < 0.001
 Fig. 6.
Fig. 6.
Schematic model. Schematic model showing how SKP2 overexpression compromised the bromocriptine-induced apoptosis by inhibiting the p53 pathway and the inhibition of SKP2 by C25 sensitizes the bromocriptine-induced apoptosis in PRL-secreting pituitary cells. SKP2, S-phase kinase associated protein 2; PRL, prolactin.
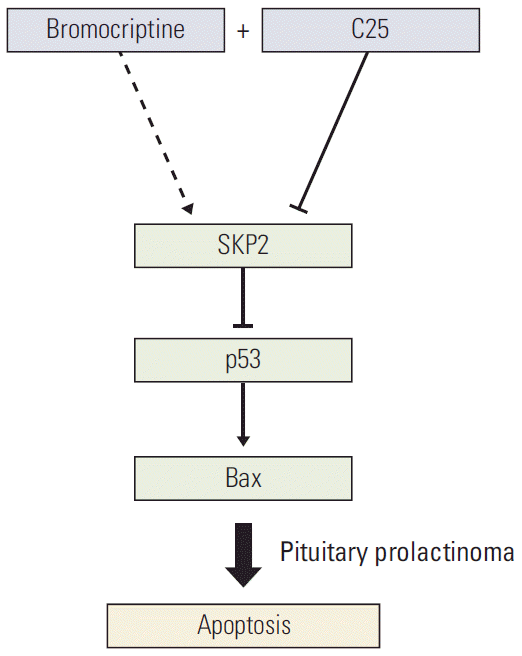 Table 1.
Table 1.
Clinical characteristics of the pituitary adenoma samples
Pituitary adenoma subtype GH PRL NFPA ACTH Knosp score 0 4 (21.1) 1 (4.3) 2 (7.7) 1 (7.7) 1 3 (15.8) 0 6 (23.1) 0 2 5 (26.3) 1 (4.3) 8 (30.8) 1 (7.7) 3 3 (15.8) 2 (8.7) 6 (23.1) 6 (46.1) 4 4 (21.1) 19 (82.6) 4 (15.4) 5 (38.5) Hardy-Wilson grade I 8 (42.1) 3 (13) 1 (3.8) 2 (15.4) II 4 (21.1) 16 (69.6) 6 (23.1) 4 (30.8) III 2 (10.5) 4 (17.4) 10 (38.5) 5 (38.5) IV 5 (26.3) 0 9 (34.6) 2 (15.4) Tumor volume (cm3) < 1 1 (5.3) 0 3 (11.5) 2 (15.4) ≥ 1 and < 2 6 (31.6) 7 (30.4) 6 (23.1) 3 (23.1) ≥ 2 and < 3 5 (26.3) 11 (47.8) 3 (11.5) 5 (38.5) ≥ 3 7 (36.8) 5 (21.7) 14 (53.8) 3 (23.1) Sex Male 7 (36.8) 3 (13) 18 (69.2) 8 (61.5) Female 12 (63.2) 20 (87.0) 8 (30.8) 5 (38.5) Age (yr) 30-40 2 (10.5) 3 (13) 5 (19.2) 0 40-50 4 (21.1) 5 (21.7) 5 (19.2) 4 (30.8) 50-60 8 (42.1) 8 (34.8) 5 (19.2) 7 (53.8) 60-70 5 (26.3)
留言 (0)