
記住我

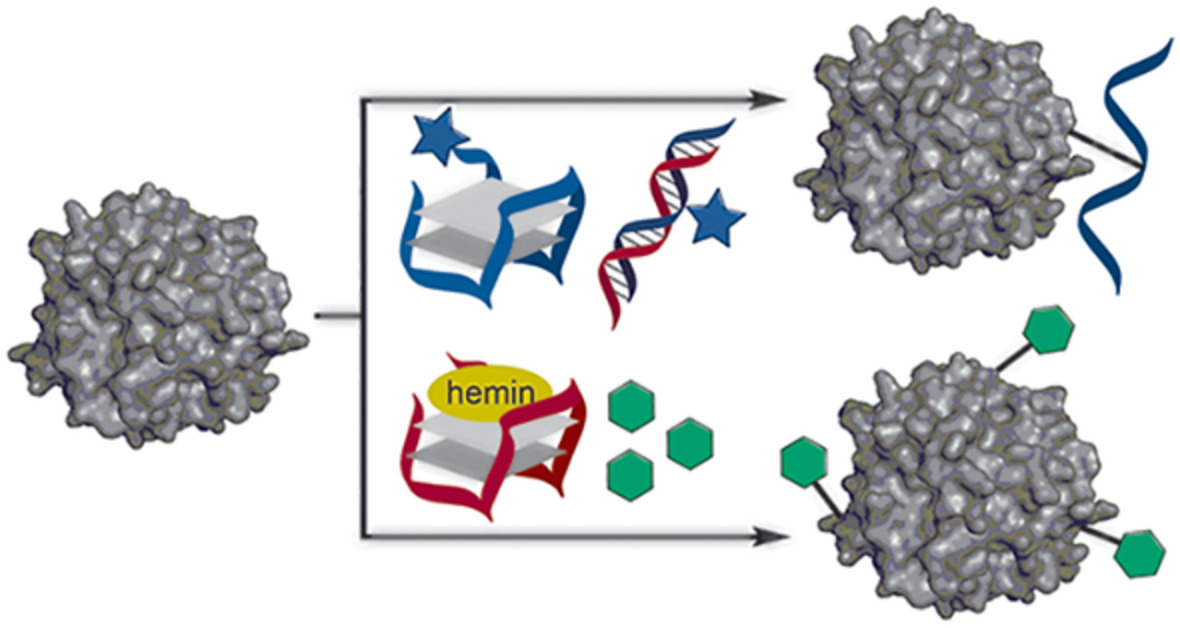



Protein modification has become a valuable tool in different fields of biochemical research, most notably in proteomics,[1] biomaterials,[2] and therapeutics.[3] Specific examples include the usage of fluorogenic protein labels to help understand intracellular protein function,[4, 5] sensors with surface-bound horse-radish peroxidase enzyme to quantify hydrogen peroxide concentrations,[6] or the application of antibodies-drug conjugates that carry toxins to specific tissues.[7, 8]
In principle, protein modification can be straightforward by exposing a protein to an electrophilic agent. For example, an N-hydroxy succinimide ester will result in multiple covalent modifications of sufficiently exposed nucleophilic residues on a protein. Unfortunately, the resulting heterogeneously modified proteins have limited high-end applications as only one (or a few) of the derivatives display(s) the desired functions. Therefore, to modify proteins in a controlled manner to obtain homogeneous conjugates, multiple methods have been developed over the past few decades.[9, 10] One of the dominant strategies today is genetic engineering of a protein to incorporate (un)natural handles (e.g., azides, olefins, alkynes) in the primary structure of proteins, or to include exposed or rare reactive residues in accessible areas of the tertiary structure (e.g., Tyr, Cys).[7, 8, 11, 12] Regrettably, genetic alteration is laborious and yields vary on a case-by-case situation.[9, 10, 13] Alternatively, optimization of the reagent to react selectively with single protein residues is possible, and research in this direction has led to sulfonyl acrylates to selective target single Lys residues,[14] and alkylbenzothiophenium to target Cys residues.[15] However, identification of such optimized reagents is again a lengthy and labor-intensive process.[9, 10, 14] As an alternative to these extensive optimization approaches, catalysts can be applied to confer the desired selectivity.[16] Known catalysts used for protein modification are the inorganic dirhodium[17] and ruthenium bipyridine complexes,[18] the organic PyOx[19] moiety, and even enzymes such as mushroom tyrosinase[7] and sortase.[20]
For many other protein modification approaches, an additional element is used to achieve a higher level of site selectivity. This element, which is often a known ligand or inhibitor for the protein of interest,[21-24] displays affinity for the protein and directs the reactive moiety -whether reactive by itself or activated by a catalyst- to a specific site on the protein. Of the ligands that have been applied, those that are based on biological entities have shown tremendous promise.[10, 16]
In this review, we discuss the methodologies developed in the recent past that harnessed the ability of DNA to facilitate, guide or even catalyze protein modification. The use of DNA has the advantage that it can be easily produced by solid-phase synthesis,[25] is relatively stable,[26, 27] is a highly programmable biopolymer that enables the design of complex supramolecular structures,[28] and in some cases can exert catalytic properties in the presence of a suitable “cofactor.”[29, 30] As such, the use of DNA for directed protein modification has great potential and already includes DNA-templated reactions, DNA-based guidance systems, and catalytic DNA constructs that enhance reaction rates. Moreover, complex systems composed of multiple DNA strands are realized by the fact that matching strands of DNA can form the typical double helix DNA, even at picomolar concentrations.[31] This concept has been exploited to enhance intermolecular reaction rates of molecules bound to the proximal ends of complementary strands,[32] but also for the modification of proteins.
2 PROTEIN MODIFICATION BY DNA TEMPLATIONThe first reported use of “DNA templation” for protein modification was by Li et al.[33] and involved various small molecules as recognition elements to guide DNA template strands (DNAtemp) to the surface of their protein binding partners (Figure 1). Afterwards, a second reacting DNA strand (DNAreact), carrying a photo-activatable diazirine moiety, was hybridized with the DNAtemp, positioning the reactive moiety in close proximity to the protein surface. Upon irradiation, crosslinking of DNAreact and the protein occurred, leading to a covalent protein-DNA conjugate. They demonstrated that this approach was compatible with multiple different small molecules, even in the presence of competitive assay conditions, such as HeLa cell lysate. Later, this method was used as a tool to identify protein targets of a DNA-encoded small molecule library.[34] After incubation of the library with cell lysate, conjugation was induced using UV light. Residual unbound DNA strands were digested by Exonuclease I, leaving only the undigested protein-dsDNA conjugates. Analysis of the remaining strands enabled the identification of both the ligand and the target protein.

DNA-templated protein modification, where the template strand (DNAtemp) is guided by different moieties (red hand). The reacting strand (DNAreact) hybridizes to the template and follow-up proximal conjugation results in site-selective attachment of DNAreact to the protein. Different guiding moieties have been adopted to target-specific proteins or protein classes using a variety of reactive groups (blue star)
A comparable DNA-templated protein modification strategy was adopted by Rosen et al.,[35] and Kodal et al.[36] who used metal-affinity probes to selectively modify His6-tagged and metalloproteins (Figure 1). A DNAtemp strand conjugated with the known chelating agent tris(nitrilotriacetic acid) (NTA) formed a complex with the His6-tag of various proteins in the presence of nickel(II) or copper(II) ions. A DNAreact strand functionalized with an N-hydroxysuccinimide was then hybridized to the protein-bound NTA-DNAtemp, enabling subsequent covalent coupling of the complementary strand to the protein target.[35] This method was not only applicable to His6-tagged protein, but also on metalloproteins and even on an IgG antibody. Further analyses of the modified proteins revealed that conjugation occurred mainly in the vicinity of metal-binding sites, making this a site-selective conjugation method. In later work, the DNA-protein conjugate was used as an intermediate, which could be oxidatively cleaved to leave an aldehyde on the protein surface. As such, the strategy uses DNA to site selectively install aldehyde groups on proteins which could subsequently be used for oxime ligation.[36]
A third strategy in this category of DNA-templated protein modification uses peptides to guide a hybridized reactive DNA strand to a specific protein (Figure 1).[37, 38] Using a DNAtemp that contained a trimethylated histone H3 peptide as a guiding moiety, proteins reading histone modification could be selectively bound within complex protein mixtures.[37] Then, the diazirine-bearing DNAreact was hybridized. Subsequent irradiation yielded the desired conjugate, which could be fished out and identified. Nielsen et al.[38] performed a similar strategy by attaching a DNAtemp to Fc-III, a cyclic peptide that is a known to bind the Fc region of human IgG.[39] After hybridization, nucleophilic attack from a lysine residue on the aldehyde of DNAreact led to the formation of an imine, which could be reductively aminated. For the antibody Rixtuximab, this resulted in 75% of DNAreact being selectively conjugated to its Fc region. In addition, the obtained conjugate was assembled into pentameric IgG superstructures, using a star-shaped DNA nanostructure as a core, to synthetically mimic IgM antibodies.[38, 40]
3 PROTEIN MODIFICATION BY DNA SUBSTRATESThe interaction between DNA and proteins has also been exploited to modify DNA-binding proteins or enzymes that have DNA as a substrate. A self-conjugating dsDNA probe was used by Dezhurov et al.[41] to label active DNA polymerase β (Figure 2a). A photosensitizer incorporated into the DNA polymerase β was triggered by irradiation and activated the 4-azido-2,3,5,6-tetrafluorophenyl group on DNAreact of the dsDNA probe, resulting in conjugation of the proximal DNA strand and trapping the probe in the DNA-polymerase active site. Even though the reported 50% conversions included unwanted products, the desired conjugates could be attained. Similarly, Liu et al.[42] used a half-dsDNA/ssDNA probe construct for the targeting of dsDNA-binding proteins, including nuclear factor-κ-β (NF-κB) (Figure 2b). The substrate part of the probe construct was used for DNA-templated protein modification (vide supra) in which a DNAreact strand that contained a diazirine moiety was hybridized to the probe. After photo-activation, the probe strand was selectively conjugated to the protein target. The system-enabled conjugation of DNA to various transcription factors and showed selectivity for dsDNA binding proteins when used in cell lysate.
 DNA-guided protein modification. (a) A photosensitizer (the light bulb) was built into a DNA polymerase to trigger a photo-activatable group on a dsDNA probe. This results in the covalent trapping of the DNA probe in the active site.[41] (b) A dsDNA probe with an ssDNA extension is used to bind the dsDNA-binding protein NF-κB. After hybridization with a diazirine-bearing DNAreact strand, photo-activation results in covalent attachment of DNAreact to NF-κB[42]
4 PROTEIN MODIFICATION BY DNA APTAMERS
DNA-guided protein modification. (a) A photosensitizer (the light bulb) was built into a DNA polymerase to trigger a photo-activatable group on a dsDNA probe. This results in the covalent trapping of the DNA probe in the active site.[41] (b) A dsDNA probe with an ssDNA extension is used to bind the dsDNA-binding protein NF-κB. After hybridization with a diazirine-bearing DNAreact strand, photo-activation results in covalent attachment of DNAreact to NF-κB[42]
4 PROTEIN MODIFICATION BY DNA APTAMERS
Although the previously mentioned works already present DNA-guided protein modification, the methods are limited to DNA-binding proteins and are not always protein specific. Alternatively, DNA-guided modification relying on the affinity of aptamers for proteins was employed. Aptamers are oligonucleotide sequences—DNA, RNA, synthetic, or hybrid—that non-covalently bind to a variety of targets, ranging from small molecules and metal ions, to large proteins and even cells.[43] This versatility in targets that can be addressed enabled aptamers to become widely used and serve as selective tools to benefit research varying from proteomics studies to therapeutic applications.[44, 45] Aptamers are discovered in a high-throughput methodology called SELEX, where a library of DNA sequences is incubated with a target, washing away nonbinding sequences and cloning the binding ones by means of PCR.[45, 46] After multiple cycles, isolation of the remaining sequences can lead to the discovery of one or more aptamers.[45, 46]
Even though aptamers can have high affinity for proteins, their binding mode not always favors efficient protein modification. To counter this, Smith et al.[47] incorporated the unnatural nucleic acid 5-bromo-2′-deoxyuridine into aptameric sequences for various proteins, among others human α-thrombin. These reactive aptamers could bind their protein target and then be covalently linked upon irradiation (Figure 3a). This approach particularly increased the capture yield of sequences with a low affinity.
 Protein modification strategies using aptamers. (a) Aptamer-Based Affinity Labeling (ABAL) uses DNA aptamers that contain reactive groups to self-conjugate after finding their target. Panel shows the reactive moieties used for ABAL and their references. (b) DNA aptamers used for DNA-templated protein modification in which DNAreact contains a photo-activatable diazirine,[52] electrophilic NHS ester[53] or an aldehyde.[54] (c) DNA aptamers tethered with one of two acyl transfer catalysts (i.e., DMAP or PyOx, see panel) that enhance acylation of thrombin with a degree of site selectivity. This system could also be switched between ON/OFF by means of an external (DNA-based) trigger[55]
Protein modification strategies using aptamers. (a) Aptamer-Based Affinity Labeling (ABAL) uses DNA aptamers that contain reactive groups to self-conjugate after finding their target. Panel shows the reactive moieties used for ABAL and their references. (b) DNA aptamers used for DNA-templated protein modification in which DNAreact contains a photo-activatable diazirine,[52] electrophilic NHS ester[53] or an aldehyde.[54] (c) DNA aptamers tethered with one of two acyl transfer catalysts (i.e., DMAP or PyOx, see panel) that enhance acylation of thrombin with a degree of site selectivity. This system could also be switched between ON/OFF by means of an external (DNA-based) trigger[55]
Vinkenborg et al.[48] developed a strategy called Aptamer-Based Affinity Labeling (ABAL), in which DNA/RNA aptamers bearing reactive moieties were used to trap their protein targets (Figure 3a). Aptamers for hepatocyte growth factor receptor, IgE, and cytohesin-2 were armed with photoreactive phenylazides and after incubation and irradiation were attached to their target with yields around 30%. Interestingly, the strategy was proven effective not only in complex protein mixtures, but also in vivo on non-small-cell lung carcinoma cells. A study from Rohrbach et al.[49] employed the ABAL approach using thrombin binding aptamer (TBA), a well-studied aptamer that binds exosite I of human α-thrombin, the clotting agent in human blood.[50] By attaching sulfo-N-hydroxysuccinimide to the 3′-end of TBA via a photocleavable tether, TBA was used to irreversibly occupy the fibrinogen binding site on the thrombin surface, thereby permanently inhibiting the activity of the enzyme. Irradiation of the formed conjugate severed the tether and allowed TBA to dissociate from thrombin, which restored its activity. This caging and decaging was demonstrated in a blood-plasma assay, where clotting was only detected after irradiation and subsequent release of TBA, indicating near quantitative masking of thrombin.[49]
Wang et al.[51] prepared the same thrombin-TBA conjugates using an α,α-difluoromethyl carboxyl group as reactive moiety, which specifically targets amine functionalities (Figure 3a). This moiety presented less off-target modification than an aptamer with an aldehyde, while retaining similar yields. The carboxyl group had to be suspended on a linker of eight deoxythymidine units, as they determined that shorter tethers hampered modification. The methodology was successfully applied for the modification of thrombin and platelet-derived growth factor with their respective aptamers.
5 PROTEIN MODIFICATION BY DNA APTAMER TEMPLATIONAs the aptameric sequence does not always possess the desired sequence to be attached to a reactive group immediately, aptamers have been used in a DNA-templated format as well. Bi et al.,[52] incubated an aptamer for lysozyme C with its designated protein target, but instead of having the aptamer react directly, it was extended with a template DNA sequence (Figure 3b). A diazirine-carrying DNAreact was hybridized to this template sequence and after photo-activation, conjugated to lysozyme C. It was shown that in this DNA-templated ABAL strategy, conjugation only occurred when the aptamer was present and that the reaction was selective in competitive assays against BSA, HeLa cell lysate, and even in raw chicken egg white with hardly any off-target conjugation.
Cui et al.[53] further used aptamer-guided DNA-templation by putting a template DNA strand on TBA. This extended TBA was hybridized with a DNAreact bearing an electrophilic N-hydroxysuccinimide ester and was successfully used to synthesize thrombin-DNA conjugates. The reaction achieved a conversion of 85% and after extraction from sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE), an isolated yield of 56% was obtained. Tryptic digestion analyses showed that the site of modification was limited to only two lysine residues, indicating high site selectivity of their method. They obtained comparable results with aptamer HD22, another aptamer for thrombin, which binds to exosite II. Since HD22 and TBA bind on opposite sides of the protein, they could be used together for dual labeling. Indeed, DNAreact was conjugated on either side of the protein, although only a small percentage of double conjugated DNA-thrombin product was obtained. Using the same method, DNA-protein conjugates were constructed with other aptamers as well, specifically for platelet-derived growth factor, streptavidin, and human IgG. One DNA aptamer with affinity for His6-tag was used to modify His-tagged Midkine, showing that this approach could also be used to recognize proteins with particular elements. In order to confirm that the target specificity of the aptamer was not compromised by the extensions, a competition experiment with BSA was performed and demonstrated that thrombin was still the only modified protein.[53]
Skovsgaard et al.[54] optimized strategies for the application of ABAL and DNA-templated ABAL on IgG antibodies. A hybrid DNA/RNA aptamer with specific affinity for the Fc domain was extended on the 3′-end with either a template strand or a reactive moiety. In both cases, an aldehyde functionality was subjected to reductive amination with nearby lysine residues on the antibody to create stable protein-DNA conjugates. The DNA-templated method generated conjugates of the therapeutic antibodies trastuzumab, rituximab, and cetuximab, with yields around 60%. The DNA-cetuximab conjugate later proved effective for the in vivo fluorescent labeling of MDA-MB-231 cells. When performing ABAL, it was revealed that this approach was more efficient, producing up to 90% conversion with just one equivalent of aptamer. Unfortunately, full site selectivity could not be achieved as conjugation to the light chain of the Fab domain (which is not part of the Fc domain) was also observed, in both the direct and DNA-templated ABAL.[54]
Alternative to these strategies, Keijzer et al.[55] developed aptamer-catalyst constructs to chemically modify thrombin. In this work, DMAP - an acyl transfer catalyst that activates thioesters- was tethered to TBA (Figure 3c) in order to acylate lysine residues in proximity of the protein–aptamer interface. Of the various constructs that contained DMAP at different positions of the aptamer, we found that functionalization of position T12 with our catalyst led to a 7-fold enhancement. Swapping DMAP for another acylation catalyst, that is, 4-pyridinecarbaldehyde oxime (PyOx), enabled the use of a more stable alkylated N-acyl-N-sulfonamide as an acyl donor. Indeed, the best performing resulting TBA-PyOx construct reached 90-fold increased conversions compared to the background. Importantly, thrombin acylation only occurred in proximity of aptamer–thrombin interface and led to site-selectively modified proteins. By attaching the PyOx catalyst to aptamer HD22, which binds to the opposite site of thrombin, modification on the opposite side occurred, revealing that the same protein could be acylated at different sides by using different aptamers. Lastly, the programmable nature of DNA allowed incorporation of an activity switch that turns the DNA-catalysts ON or OFF by means of an external trigger.
6 PROTEIN MODIFICATION BY DNAZYMESDNA sequences can also bind cofactors leading to a complex that can exert catalytic functions. The majority of these so-called DNAzymes have been reported only in the past two decades, the first of which was an RNA-cleaving DNAzyme that was discovered in 1994 by Breaker and Joyce[56] and since then, more DNAzymes have been reported that catalyze various reactions.[30, 57, 58] One type of DNAzyme is the horseradish peroxidase (HRP) mimicking hemin/G-Quadruplex (hGQ) DNAzyme. It is a potent oxidative catalyst,59, 60 which is formed by combining a G-quadruplex (GQ) forming oligonucleotide strand and hemin, the iron(III)protoporphyrin complex found in hemoglobin.[60] In the presence of exogenously added hydrogen peroxide, hemin can oxidize various organic substrates, but is on its own prone to autodegradation.[61, 62] The GQ secondary structure not only stabilizes hemin, but also enhances its activity, resulting in a DNA-based catalyst that can oxidize various substrates.[59-62] The hGQ DNAzyme has been known to have the potential for protein conjugation, as it can produce cross-coupling of tyrosine residues.[63] This reaction has been used for tyramide deposition on cell surface proteins,[64] but protein conjugate yields were regrettably never high.
Recently, Keijzer et al.[65] demonstrated that the hGQ DNAzyme can also catalyze the oxidative conjugation of N-methylluminol (NML) derivatives to tyrosine (and to a lesser extend tryptophan residues) on several proteins, including lysozyme C, human α-thrombin, bovine serum albumin (BSA), and the immunoglobulin trastuzumab (Figure 4).[65] Studies regarding multiple GQ-forming DNA sequences showed that the morphology of the GQ strand was of great influence on the conversions percentage. Also, different GQ morphologies led to different residues being modified, which was hypothesized to be caused by the GQ DNA binding the protein at different sites or angles. Moreover, in the presence of an aptamer for the target protein, the site selectivity of the modification shifted and a residue could be modified that was not modified in the absence of the aptamer. Notably, a G-rich DNA strand that could not adopt any GQ folding presented no amplified hemin activity. This made it possible to devise a system in which the DNA catalyst could be switched between its GQ-folded active state (ON) and a dsDNA duplex inactive system (OFF) (similar to what was shown in Figure 3c). The result was a controllable catalyst with 80% conversion in the ON state and only 5% conversion in the OFF state.[65]
 Protein modification by means of DNA-based catalysts. Using H2O2 as radical source, hGQ DNAzymes[64, 65] (and RNAzymes)[66] were used to conjugate phenols or N-methylluminol (NML) derivatives to tyrosine residues on proteins. The box shows a model of an hGQ DNAzyme
Protein modification by means of DNA-based catalysts. Using H2O2 as radical source, hGQ DNAzymes[64, 65] (and RNAzymes)[66] were used to conjugate phenols or N-methylluminol (NML) derivatives to tyrosine residues on proteins. The box shows a model of an hGQ DNAzyme
At the same time, a paper by Masuzawa et al.[66] demonstrated similar protein conjugation using RNA-based hGQ complexes. After combining the telomeric repeat-containing RNA sequence with hemin, the formed hGQ RNAzyme demonstrated capable of selectively labeling RNA-binding proteins. With yields of about 50% on known RNA-binder Unwinding Protein 1, the modification capacity was used to tag RNA-binding proteins with biotin in HeLa cell lysate and capture them by means of streptavidin beads. Proteomics studies revealed that 82 of the 480 captured conjugates were indeed RNA-binding proteins.[66]
7 CONCLUSIONS AND OUTLOOKIn conclusion, we reviewed various approaches in which DNA has been used to assist in selective protein modification (Table 1). These include DNA templation, a ligand-directed approach, protein-binding aptamer sequences, and even DNA-based catalysts. As such, DNA has proven to be an effective aid in achieving site-selective protein modification and the fruits of these works are already being put to good use.[67-69]
TABLE 1. Overview of the DNA-assisted protein modification strategies described in this review, including associated conversions, advantages, disadvantages, and appropriate references Methods Details Conversions Advantages Disadvantages ReferenceDNA-templation

small molecule
0.1–2a Small guiding unit Highly specific Many protein ligands available Limited to available ligands Increase of KD by DNA attachment [33, 34]
metal-affinity
25%–60% Applicable to His6-tagged proteins Metal binding site on protein required Metal ion required [35]
peptide
50%-100% Many protein-binding peptides available Variation in attachment point of DNA Protein-binding peptides are sometimes large Poorly defined peptide-protein interaction [36-38] DNA substrate dsDNA probe
±50%
Selective for dsDNA binding proteins
Straight forward binding probe
Requires engineering of functionalized dsDNA
Case-specific strand length optimization
Limited to dsDNA binding proteins
[41]
Templated dsDNA probe
0.1–2a
[42]
DNA aptamer as guiding unit
dsDNA probe
±50%
Selective for dsDNA binding proteins
Straight forward binding probe
Requires engineering of functionalized dsDNA
Case-specific strand length optimization
Limited to dsDNA binding proteins
[41]
Templated dsDNA probe
0.1–2a
[42]
DNA aptamer as guiding unit
 20%-100%
Protein specific
No additional binding unit required
Weak-binding aptamers also ligate
Only self-conjugate
Limited number of known aptamers
Poorly defined aptamer-protein interaction
[47-49, 51, 54]
20%-100%
Protein specific
No additional binding unit required
Weak-binding aptamers also ligate
Only self-conjugate
Limited number of known aptamers
Poorly defined aptamer-protein interaction
[47-49, 51, 54]
 45%–85%
Protein specific
Large variation on complementary strands possible
Only conjugate complementary strand
Limited number of known aptamers
Poorly defined aptamer-protein interaction
[52-54]
45%–85%
Protein specific
Large variation on complementary strands possible
Only conjugate complementary strand
Limited number of known aptamers
Poorly defined aptamer-protein interaction
[52-54]
 35%–100%
Protein specific
High conversions
Conjugate small molecules
Switchable activity
Limited number of known aptamers
Possible off-target reactivity
Poorly defined aptamer-protein interaction
[55]
DNA catalyst
35%–100%
Protein specific
High conversions
Conjugate small molecules
Switchable activity
Limited number of known aptamers
Possible off-target reactivity
Poorly defined aptamer-protein interaction
[55]
DNA catalyst DNAzymes
25%–100%
Short reaction time
High conversions
Conjugate small molecules
Switchable activity
Requires H2O2
Limited set of organic substrates
Unwanted protein oxidation
[64, 65]
RNAzymes
±50%
[66]
a
These are normalized yields.
DNAzymes
25%–100%
Short reaction time
High conversions
Conjugate small molecules
Switchable activity
Requires H2O2
Limited set of organic substrates
Unwanted protein oxidation
[64, 65]
RNAzymes
±50%
[66]
a
These are normalized yields.
Looking at the future, we realize that DNA-assisted protein modification is still in its infancy. Therefore, it is our perception that advanced DNA-based approaches have the potential to generate novel site-selective conjugation methods. Specifically, the following major benefits of DNA will aid in the development of such tools: (i) the compatibility of DNA with a variety of chemical and biological agents, (ii) the convenience by which synthetic modifications can be inc
留言 (0)