
記住我




 leads to a manifold of different conformations characterized by their relative strand orientation and the resulting intramolecular loop geometries (Figure 1II).[9]
leads to a manifold of different conformations characterized by their relative strand orientation and the resulting intramolecular loop geometries (Figure 1II).[9]
 Basic principles of G4 architecture. (I) G-tetrad hydrogen bond pattern and tetrad stacking to form the G4 core. Right: Schematic representation of a G4 with G-residues shown in green. Monovalent cations (M+) are indicated with blue spheres. (II) Canonical G4 structural polymorphism with different folding topologies. Loops I-III and G-tracts 1–4 are indicated in 5′-3′ direction. Conformations: (A) hybrid (3 + 1) with edgewise (blue) and double chain-reversal (red) loops; (B) anti-parallel (2 + 2, basket) with diagonal (green) loop; (C) parallel. (III) Non-canonical polymorphism with (d) G-vacancy site, (e) bulged nucleotide and (f) snap-back motif. (IV) Forms of non-canonical folding isomerism with (g) G-register shifts and (h,i) spare-tire exchange (Figure parts adapted from Grün[8])
Basic principles of G4 architecture. (I) G-tetrad hydrogen bond pattern and tetrad stacking to form the G4 core. Right: Schematic representation of a G4 with G-residues shown in green. Monovalent cations (M+) are indicated with blue spheres. (II) Canonical G4 structural polymorphism with different folding topologies. Loops I-III and G-tracts 1–4 are indicated in 5′-3′ direction. Conformations: (A) hybrid (3 + 1) with edgewise (blue) and double chain-reversal (red) loops; (B) anti-parallel (2 + 2, basket) with diagonal (green) loop; (C) parallel. (III) Non-canonical polymorphism with (d) G-vacancy site, (e) bulged nucleotide and (f) snap-back motif. (IV) Forms of non-canonical folding isomerism with (g) G-register shifts and (h,i) spare-tire exchange (Figure parts adapted from Grün[8])
A sequence-based prediction for distinct conformations is already complex within this canonical set of G4 topologies.[10] In addition to this canonical G4 structural polymorphism, new aspects of structural complexity have been described recently, which are referred to as non-canonical polymorphism.[2, 11-13] Aspects including bulges,[14-16] exceptional loop arrangements[17, 18] and snap-back motifs[19-23] can be observed in G4s from sequences that do not comply with the narrow definition given above (Figure 1, III).
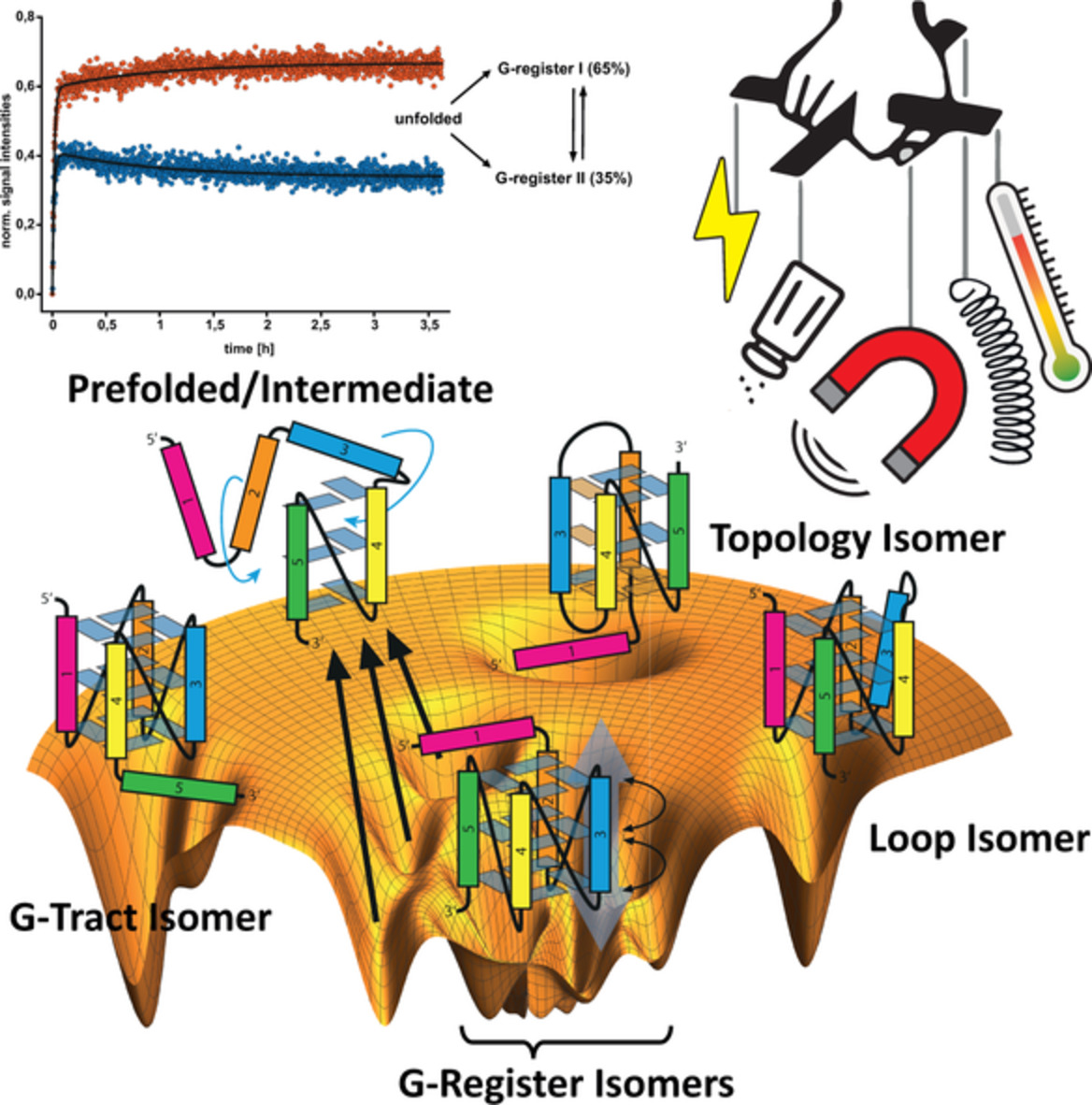
Polymorphism is pronounced among G4s from different sequences, but is also observed within a given G4 forming sequence, leading to concurrent folding isomers in heterogenous ensembles.[24-26] In an enlarged conformational space, due to additional combinatorial possibilities, two particularly intriguing forms of folding isomerism arise (Figure 1, IV):
Firstly, if the number of subsequent G-residues exceeds (or undercuts[27-29]) the number of G-tetrad layers in a quadruplex, an exchange of a single G-register along the G4 core can be observed, leading to a conformational subset of shifted G-register isomers.[30]
Secondly, if the number of subsequent G-tracts is greater than four, different isomers can be formed by incorporating different G-tracts into the formation of the G4 core.[31, 32] For the latter, the term spare-tire isomerism has been newly coined.[31, 33]
Note that we here define both forms of folding isomerism with respect to the relation of the involved distinct G4 conformations arising from the same nucleotide sequence.
1.2 Regulatory role of G4The emerging role of G4 forming elements in the human genome has been reviewed extensively in the context of transcription regulation,[34] replication,[35, 36] genomic instability,[37, 38] epigenetic modifications[39-43] and telomer stabilization.[44] Since the first reports on transcription regulation through small-molecule ligands that target gene promoter G4s,[45] a plethora of approaches have evolved that focus on the development and characterization of G4 stabilizing agents.[46, 47] While these strategies aim at stabilizing G4s as molecular mechanism in novel anti-cancer therapies,[48] more recently, converse strategies have been proposed to counteract genomic instability induced by the stable formation of G4s in chromosomal DNA.[37] Both these general approaches, stabilization and destabilization, interfere with a misregulated G4 formation at a pathological stage. The ambivalent consequences of G4 structure formation in different contexts highlights the requirement for the dynamic regulation of G4s: transient folding and unfolding has to be maintained for balanced cellular homeostasis. It is thus not surprising that an inherently dynamic nature is also a key feature of G4 formation in RNAs.[49-51]
The versatile potential to fold into a manifold of distinct G4 structures has thermodynamic, kinetic and also biological consequences. In particular for the latter, it is a crucial aspect to ensure adaptability of G4 elements in response to external stimuli. As a prime example for the structural adaptability, the functional role of spare-tire G-tracts has been proposed to maintain G4 formation after oxidative damage, since G-stretches with increasing lengths are especially prone to oxidize.[31, 32, 52-55] This mechanism enables the subsequent recruitment of repair machineries, which can reset the damaged DNA stretch.[55, 56] In this simple picture, the maintenance of function for polymorphic G4s refers to an on/off switching for example, in transcriptional control, if a G4 structure is present or not.
However, there is now growing evidence that G4 polymorphism itself is a crucial aspect of G4 regulatory function. We exemplify these new findings with two structural aspects: Spare-tire isomers with different loop lengths have drastically different affinities towards G4 interacting proteins, even for G4s with the same folding topology (e.g., parallel loop isomers).[57-59] Structural isomers with different topologies (e.g., hybrid and parallel) result in vastly different unwinding efficiencies for G4 specific helicases.[38, 60-65] These structural aspects might already arise from small changes. Thus, while formation and fold topology of a G4 might be maintained in principle, the result of modified sequences (due to oxidation or mutations[66, 67]) could be a completely altered regulation cascade.[59]
In view of the consequences of G4 polymorphism, the simple model that G4s act as steric bulking structures, often anthropomorphically called roadblocks, is not suited to explain their regulatory function.[39] Hence, statements about a general functionality for G4 formation have to be taken with care, whenever the specific conformation is not considered. For G4 forming sequences that are able to fold into different stable conformations, the individual kinetics of concurrent folding pathways towards a specific conformation might be biologically even more relevant than the stabilities at thermal equilibrium.
2 EXPERIMENTAL ASPECTS 2.1 Preparation of non-equilibrium conformational statesA prerequisite to study coherent structural changes is the preparation of suitable starting points away from equilibrium (Figure 2). The experimental approaches to prepare any kind of trapped, retained or excited state can heavily influence the folding progression, possible pathways and kinetics.[68-70] It is thus worthwhile to compare and evaluate the experimental premises to understand possible ambiguous results.

Non-equilibrium G4 dynamics. (I) Reversible methods: (A) mechanical unwinding with magnetic/optic beads, (B) thermal melting observed with T-jump or thermal hysteresis. (II) Irreversible folding methods: (C) rapid mixing with monovalent cations, (D) photolytic release of photolabile protecting groups (photocages). (III, E) conformational selection of folded G4s with photocages, after photolysis complete refolding or re-equilibration into a polymorphic ensemble [*] can be observed. (IV) Irreversible Unfolding methods: (F) Photocleavage of the DNA backbone to destabilize intramolecular G4s, (G) complement trapping to induce duplex formation
2.1.1 Reversible folding (I) Mechanical unwindingMechanical unwinding is an intriguing, but technically demanding possibility to investigate G4 folding as a measure of force under isothermal experimental conditions and at constant cation concentration (Figure 2A). Cheng et al. have used this method to study the folding/unfolding of a BCL2 promoter G4 with single molecule force spectroscopy; the observed force changes are in the range of pN.[71] Using a tethered oligonucleotide that has been fixed with magnetic beads, they were able to describe kinetic differences for spare-tire isomers of the BCL2 G4 with this approach. While this method clearly ensures a pure unfolded state, it should be noted that this state will be characterized by inherently lower conformational entropy compared to other denatured states due to fewer translational degrees of freedom.[72]
Thermal hysteresisIn reversible thermal melting and annealing experiments, G4 forming oligonucleotides can show pronounced hysteresis (Figure 2B).[73-75] The complex behavior at thermal transitions can be used to gain insight into the folding process of G4s, typically with photometric detection (UV/CD). Mittermaier et al. have presented a sophisticated strategy to extract dynamic information from experiments under deliberately chosen conditions that provoke thermal hysteresis.[30, 69, 73, 76] Rapid temperature jumps (T-jump) have been used to induce G4 folding in circular dichroism (CD)-spectroscopic[77] and mass spectrometric[78] setups and new probe designs could potentially allow T-jump induced folding also for NMR spectroscopy.[79-81] Thermal (un-)folding is typically limited to lower than physiological K+ concentrations, due to the high thermal stability of many G4 structures.[74, 75]
2.1.2 Irreversible folding (II) and refolding (III) Cation-induced foldingA widespread strategy of inducing G4 folding is to dissolve DNA with G4 forming sequences under buffer conditions that lack G4 stabilizing cations (Figure 2C). Thus, G4 folding is inhibited at ambient temperatures. Under careful experimental control (e.g., unwanted cation uptake from tubes), in particular DNA G4s can be prepared in an unfolded state. Folding at a specific temperature then can be induced by mixing with for example, Na+ or K+ which allows the application for any spectroscopical method as readout.[82] In general, cation-induced folding is a very simple, reliable and broadly applicable method, yet the degree of denaturation has to be evaluated carefully with spectroscopic methods. For RNA G4s for example, preparation of an unfolded state following this procedure is more difficult. In a previous study pre-formation of an RNA G4 fold was observed even in the obvious absence of K+-ions and complete folding occurs at much lower K+ equivalents as compared to the corresponding DNA sequence.[83] CD spectra provide characteristic patterns for specific G4 architectures. Even more insightful, NMR spectra of the fingerprint region show hydrogen-bonded imino 1H signals that allow a sensitive evaluation of residual structure formation.[84] Indeed, in many cases the formation of pre-folded states (see below) has been observed or structure formation even at very low, sub stoichiometric K+ concentrations.[83, 85] In many experiments, the concentration of K+ is often below physiological concentrations, which greatly affects the thermal stability of G4s.[75] The kinetics are accelerated with increasing K+ concentration (even at unphysiological high concentrations of greater than 100 mM)[86] but the main effects, in particular the branching of pathways are present already at very low K+ concentrations (<3 mM).[82, 83]
Photocaging for conformational selectionPhotocaging of RNA and DNA with photolabile protecting groups on their nucleobase moieties can be applied for studying G4 folding.[87, 88] Photocages can inhibit hydrogen bond interactions site specifically on distinct nucleobases in the oligonucleotide and can be removed upon light irradiation with a selective wavelength, thereby releasing the completely unmodified nucleobase. This concept was first exploited to study RNA refolding,[89-92] and has been applied to G4s recently in two different ways, either by selecting single folded conformations out of a polymorphic conformational ensemble from a G4 forming sequence instead of using sequence mutations (Figure 2E)[69]; or to suppress completely the G4 folding to trap the unfolded state (Figure 2D).[33, 76] The method allows investigating the isothermal folding at constant experimental conditions and ensures a robust disruption of hydrogen bond interactions or preferential pre-orientations. While photocages are introduced at the nucleobases and act irreversible in the direction of folding, the incorporation of photosensitive scaffolds into the DNA backbone can be used to cleave and hence unfold G4s (Figure 2F),[93] or enable a reversible switching between different G4 conformational states.[94]
2.1.3 Irreversible unfolding (IV) Unfolding with complementary trappingThe folded state itself can also serve as starting point for investigating the reverse folding trajectory of an order–disorder transition (Figure 2G). Addition of the complementary strand (complementary trapping) has been used with PNA and DNA strands to trigger the irreversible unfolding of G4s towards a duplex fold.[95-98] The observed kinetic rates for unfolding in these experiments are typically orders of magnitude slower than for folding, reflecting the high stability of the G4 fold. Klejevskaja et al. have studied a self-assembled double stranded DNA mini-circle with an embedded single stranded region that codes for the cMYC G4.[99] Using a clever FRET-based strategy with two dyes placed at the brink of the inserted G4 element, they measured the G4 unfolding kinetics after adding the complementary strand. The observed unfolding is approximately 10 times slower than for the isolated single stranded G4 oligonucleotide, reflecting a higher kinetic stability under conditions with restricted flexibility that resemble the chromosomal context of genomic DNA. Since the complementary strand itself is typically able to form a folded i-motif secondary structure, the experimental conditions for complement trapping have to be evaluated carefully to prevent convolution of the structural dynamics.
2.2 Spectroscopic methods to study folding and refolding kinetics 2.2.1 Circular dichroismCD spectroscopy is an excellent method to monitor folding kinetics of G4s and provides an easy access to the basic structural constitution of G4s. Much of the pioneering work on G4 folding has been conducted or supported by CD spectroscopy.[75, 82, 95, 100-102] CD spectroscopy gives a simple and characteristic readout that can be used to distinguish between an unfolded state and different folded conformations [parallel: ~260-265 nm (+), anti-parallel: ~295 nm (+), hybrid ~265/295 nm (+)].[103] CD spectroscopy is mainly indifferent to aspects of non-canonical polymorphism such as parallel G-register shifted or spare-tire isomers. However, there are now sophisticated analysis tools available that allow deconvolution of complex CD spectra from polymorphic G4s.[103, 104] Time-resolved CD spectroscopy was used in combination with an laser-induced temperature jump to monitor G4 folding down to a millisecond timescale.[77] UV spectroscopy is suited as a readout in a similar way, but is restricted to a simple “folded/non-folded” monitoring with characteristic changes at 295 nm.[105-108]
2.2.2 Mass spectrometryMass spectrometry (MS) gives orthogonal insight into aspects of G4 folding that are not directly available with spectroscopy, while MS itself does not provide direct structural information.[109] MS has been used for the evaluation of G4-ligand binding and of the folding pathways of G4s by analysis of cation binding stoichiometry.[110, 111] A precise evaluation of K+ binding to the DNA strand is crucial to understand the enthalpic and entropic contributions that affect G4 folding in thermal experiments.[111] In a recent fascinating paper, Gabelica et al. have presented an approach for the detection of mass-resolved CD spectra of G4 forming oligonucleotides.[112] This powerful method in combination with advanced computational methods for the deconvolution of CD spectroscopical and mass spectrometric parameters will enable new perspectives on G4 folding.[104, 113-116]
2.2.3 Single molecule spectroscopyForce spectroscopy/microscopy can be used for directed force manipulations on G4 oligonucleotides accomplished with magnetic[71, 117-119] or optical beads/tweezers,[120-122] also in combination with fluorescence detection.[123-125] Sugiyama et al. have demonstrated the observation of G4 folding in DNA nanostructures using high-speed atomic force microscopy (AFM).[72, 126-128] Förster resonance electron transfer (FRET) yields a very specific readout for two-site distances, which requires, however, the incorporation of dye labels that potentially bias the G4 structural integrity.[75, 129] While FRET provides no direct information on the G4 conformation it allows a very selective observation of folding trajectories in single molecule experiments.[99, 130, 131] The selective observation of different FRET states make this method suitable for the application in high molecular weight complexes, in particular for the investigation of G4 unwinding by helicases.[64, 65, 132-134]
2.2.4 Nuclear magnetic resonanceNuclear magnetic resonance (NMR) spectroscopy is a powerful and versatile method to study G4 structural dynamics at atomic resolution.[81, 135] Substantial information on the number of states adopted by a given G4 can already be read-off in one-dimensional NMR spectra, as the spectral regions for imino hydrogen atoms involved in Watson-Crick, Hoogsteen or i-motif interactions are clearly distinct. Counting the number of resolved imino hydrogen atoms often already provides a direct readout of multiple, polymorphic states in slow conformational exchange, implying at least millisecond lifetimes of these states.[136] The quantification of arising imino 1H signals in time-resolved experiments was used to study the folding of RNA and DNA G4s[33, 69, 83, 85] and DNA i-motifs,[137] respectively. To access the rich and complex structural information of NMR spectra, however, higher dimensional homo- or heteronuclear correlated spectra are required since signal resolution decreases with increasing molecular size.[138-140] Especially nucleic acids show an inherently poor spectral dispersion due to only four different nucleobases that constitute the basic polymer building blocks.[136]
3 FOLDING ENERGY LANDSCAPE AND FOLDING PATHWAYS 3.1 Ensemble effectsFrom NMR and CD spectroscopic G4 folding experiments, we derive a conformational energy landscape that depicts the entire experimentally observable conformational space of G4 DNAs. To some extent, this landscape is simplified, compared to theoretical landscapes predicted by molecular dynamics (MD) simulations that potentially aim to represent the complete phase space.[70, 141] In spectroscopic experiments only macrostates, represented by a particular conformational state (or structural fold) of the DNA strand of a certain lifetime can be observed. The multiples of subordinated microstates that contribute to for example, the conformational entropy of a macrostate thus are rather a subject of MD simulations than experimental evidence. Nevertheless, also in experimentally derived kinetic models conformational states should be referred to as ensembles (e.g., transitory ensemble), when the range of involved microstates exceeds a structurally clearly defined macrostate.[142]
The description of folding pathways can be experimentally achieved on a single molecule level or in an ensemble average. In NMR, for example, the observation of an ergodic ensemble[143] (100 μM NMR sample ≈ 300 nmol DNA ≈ 1017 folding events) leads to an extensive mapping of the energy landscape. In CD spectroscopy, this number is lower by a factor not smaller than 10−3. Different to other biomolecules, in the special case of chromosomal DNAs the folding of a particular G4 sequence is not an ensemble process, but a single event in each living cell. The assumption that G4 folding is an infrequent event is based on the fact that G4 folding does not happen spontaneously in double stranded DNA. A presumable requirement for G4 folding in any chromosomal region different from the single stranded telomeres is negative superhelicity, which is induced during transcription or replication.[39, 144-147] Interestingly, G4 folding is also associated to accessible chromatin states and therefore might even precede transcription.[148] Regardless of other cellular triggers for G4 folding, we try to give a rough estimate for the relevant rates of G4 folding events with respect to transcription. Assuming a total intracellular concentration of ~105 mRNAs per cell,[149, 150] with a median copy number per gene of ~17 per mRNA (in comparison: the protein concentration is ~109 per cell,[151] with ~50.000 copies per protein[152, 153]). The typical intracellular lifetimes for mRNAs are longer than several hours,[152-154] but especially regulatory mRNAs have significantly shorter lifetimes,[154] leading to a potentially higher transcription rate of certain G4 mediated genes. However, these calculations still lead to only a very few, approx. <100 potential folding events of a distinct promoter G4 per hour per cell. Hence, the total number of transcription initiated G4 folding events is hardly to call a dynamic ensemble. This situation changes, if tissues are considered: in a tumor tissue for example, ~108 to ~109 cells are observed per gram tissues,[155] which adds up to a tremendous number of independent G4 folding processes in vivo.
3.2 A view from computationIn stark contrast to funnel-like energy landscapes that describe folding trajectories of proteins, G4-forming oligonucleotides exhibit a rough conformational energy landscape (Figure 3, I).[70, 141] Instead of approaching a native folded state following a funnel-like, two-state folding transition, different competing basins of metastable states are observed. These basins, or flat wells, in many instances lead to concurrently, coexisting folded conformations (different macrostates) in thermal equilibrium. During folding, during which high-energy non-equilibrium states are initially formed, different conformational states with low energy barriers can be sampled. Thus, the routes along the folding energy landscapes are rather complex and involve stochastic sampling of different folding and misfolding pathways. The relative contributions of different pathways for folding reaction undergoing kinetics partitioning has been studied with MD simulations, and revealed possible involvements of hairpins,[142, 156] triplexes[157] and strand slipped conformations.[29, 70, 158] Stable G-hairpins,[159] cross or parallel G-hairpins[142] and newly discovered pseudocircular G-hairpins[160] represent interesting possible waymarks along the folding pathways. Derived from the computational picture of the conformational energy landscape, the underlying molecular folding mechanisms with competing trajectories are described as kinetic partitioning mechanism, as opposed to a funnel-like mechanism.[70]
 Folding Pathways along the rough conformational energy landscape. (I) A central native basin of attraction represents an ensemble of conformational states with similar stabilities and low relative energy barriers. Competing basins of attraction might trap conformations that are separated by high activation energy barriers. (A) Folding into a competing basin can result in isolated conformations (coexisting or metastable). (B) Multiphasic folding pathway with short-lived intermediates. (C) Folding into a competing basin that can refold into the native basin. (D) Refolding via transitory ensembles with high activation energy barriers. (E) Parallel folding pathways with direct folding (1) and folding via off-pathway intermediates that require refolding (2). (F) Low activation energy barrier refolding or minor rearrangements between coexisting states. (II) Examples for experimental observations of kinetic partitioning folding mechanisms; kinetic traces and proposed kinetic models for different conformations of the cMYC G4. (G) Folding with parallel pathways and off-pathway formation according to (E). (H) Folding in two coexisting G-register isomers with subsequent refolding according to (B,F) (Figure parts adapted from Grün[8])
3.3 Folding kinetics and rate constants
Folding Pathways along the rough conformational energy landscape. (I) A central native basin of attraction represents an ensemble of conformational states with similar stabilities and low relative energy barriers. Competing basins of attraction might trap conformations that are separated by high activation energy barriers. (A) Folding into a competing basin can result in isolated conformations (coexisting or metastable). (B) Multiphasic folding pathway with short-lived intermediates. (C) Folding into a competing basin that can refold into the native basin. (D) Refolding via transitory ensembles with high activation energy barriers. (E) Parallel folding pathways with direct folding (1) and folding via off-pathway intermediates that require refolding (2). (F) Low activation energy barrier refolding or minor rearrangements between coexisting states. (II) Examples for experimental observations of kinetic partitioning folding mechanisms; kinetic traces and proposed kinetic models for different conformations of the cMYC G4. (G) Folding with parallel pathways and off-pathway formation according to (E). (H) Folding in two coexisting G-register isomers with subsequent refolding according to (B,F) (Figure parts adapted from Grün[8])
3.3 Folding kinetics and rate constants
The kinetic partitioning populating parallel folding pathways causes complex and multiphasic folding kinetics (Figu
留言 (0)