Trial Design and Oversight
This was a multicenter, randomized, double-blind, double-dummy, placebo-controlled trial evaluating the efficacy and safety of two abrocitinib doses in adults with moderate-to-severe atopic dermatitis who were receiving background topical therapy. Patients were enrolled in 18 countries (Australia and countries across North and South America, Europe, and Asia) from October 29, 2018, to August 5, 2019.
Patients entered a 28-day screening period, during which systemic and topical medications for atopic dermatitis were discontinued before the first dose of a trial medication or placebo was administered. Emollients were used twice daily, starting at least 7 days before randomization and continued throughout the trial, and therapy with a topical medication (applied once daily) was started on day 1 of the treatment period (baseline). Topical therapies that were allowed during the trial included low- or medium-potency topical glucocorticoids, topical calcineurin inhibitors, and topical phosphodiesterase 4 inhibitors; patients were allowed to use more than one topical agent. Rescue therapy with systemic medications or topical treatments was not permitted if it included agents that were more potent than the allowed low- or medium-potency agents. The investigators, who were unaware of the trial-group assignments, evaluated efficacy and safety by means of a telephone call at week 1 and in-person visits at weeks 2, 4, 8, 12, 16, 18, and 20. Patients who completed the trial were allowed to enter a long-term phase 3 extension trial (ClinicalTrials.gov number, NCT03422822).
The trial was conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guidelines. All local regulatory requirements were followed. This research was approved by the institutional review board or ethics committee at each trial site. All patients provided written informed consent. An external data monitoring committee reviewed safety data throughout the trial.
The trial sponsor (Pfizer) designed the trial with input from the fourth author. The sponsor collected and analyzed the data and provided the drugs and placebo. The first draft of the manuscript was written by the last author, and medical writing and editorial support with subsequent drafts (funded by the sponsor and conducted in accordance with Good Publication Practice guidelines) was provided by ApotheCom, a medical communications company, under the guidance of the authors. The analysis of the data was performed by two authors employed by the sponsor and by the sponsor’s programming group. All the authors vouch for the accuracy and completeness of the data, for the complete reporting of adverse events, and for the fidelity of the trial to the protocol (available with the full text of this article at NEJM.org). Confidentiality agreements were in place between the authors and the sponsor.
Patients
Patients were eligible to participate if they were 18 years of age or older and had at least a 1-year history of atopic dermatitis that was moderate to severe at baseline, as determined by a score of 3 or higher on the Investigator’s Global Assessment (IGA; scored on a 5-point scale [0, clear; 1, almost clear; 2, mild; 3, moderate; and 4, severe])18; a score of at least 16 on the Eczema Area and Severity Index (EASI; scores range from 0 to 72, with higher scores indicating greater severity)19; at least 10% body-surface-area involvement; and a score of at least 4 on the Peak Pruritus Numerical Rating Scale (PP-NRS; scores range from 0 to 10, with higher scores indicating greater pruritus).20 The PP-NRS was used with permission from Regeneron Pharmaceuticals and Sanofi, which developed the scoring system. During the 6 months before screening, all the patients had an inadequate response to topical medications that were given for at least 4 weeks or a need for systemic therapy to control their disease. Patients who had previously used systemic JAK inhibitors or dupilumab or had a medical history of conditions associated with thrombocytopenia, coagulopathy, or platelet dysfunction were ineligible. Patients with a history of conjunctivitis or similar eye conditions were not excluded. A complete list of inclusion and exclusion criteria is provided in the protocol.
Randomization
Patients were randomly assigned in a 2:2:2:1 ratio to receive 200 mg or 100 mg of abrocitinib orally once daily, 300 mg of dupilumab subcutaneously every other week (after a loading dose of 600 mg), or placebo for 16 weeks. The patients, investigators, and representatives of the sponsor were unaware of the trial-group assignments. Additional information on dosing and concomitant topical therapies is provided in Section S3 of the Supplementary Appendix, available at NEJM.org.
End Points
The primary end points were an IGA response (defined as a score of 0 or 1 on the IGA, with an improvement of ≥2 points from baseline) and an EASI-75 response (defined as ≥75% improvement from baseline in the score on the EASI) at week 12. Both doses of abrocitinib must have been significantly better than placebo on the basis of both end points in order to have met the goal of the trial.
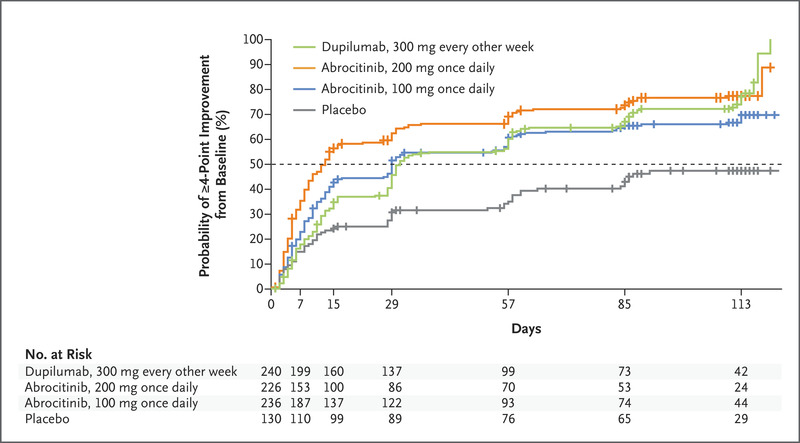
The three key secondary end points were itch response (defined as ≥4-point improvement from baseline in the score on the PP-NRS) at week 2 and IGA and EASI-75 responses at week 16. Daily scores on the PP-NRS were used in the analysis of this end point rather than average weekly scores.
Additional secondary end points included improvements of at least 50%, at least 90%, and 100% in the score on the EASI; time to itch response; changes in percentage of body-surface-area involvement; score on the Patient-Oriented Eczema Measure (POEM; scores range from 0 to 28, with higher scores indicating greater severity)21; score on the Pruritus and Symptoms Assessment for Atopic Dermatitis (PSAAD; scores range from 0 to 10, with higher scores indicating greater severity)22; score on the Dermatology Life Quality Index (DLQI; scores range from 0 to 30, with higher scores indicating greater impairment)23; scores on the Hospital Anxiety and Depression Scale (scores range from 0 to 21, with higher scores indicating greater anxiety or depression)24; improvements of at least 50% and at least 75% in the score on the Scoring of Atopic Dermatitis (SCORAD; scores range from 0 to 103, with higher scores indicating greater severity)25; and change in SCORAD subjective assessments of itch and sleep loss. A PP-NRS score lower than 2 was a post hoc end point. A complete list of the scales used in the trial end-point assessments is provided in Section S4.
At each trial site, the clinical trial investigators, who were unaware of the trial-group assignments, recorded adverse events from the time after a patient signed the informed consent through 28 days after the trial regimen was discontinued. They also recorded abnormal clinical findings and changes from baseline in clinical laboratory values, electrocardiogram measurements, and vital signs from the time after the administration of the first dose of a trial drug or placebo through 28 days after the trial regimen was discontinued.
Statistical Analysis
We determined that a sample size of 700 patients would provide the trial with at least 96% power to detect a difference of 20 or more percentage points between the abrocitinib dose groups and the placebo group with respect to an IGA response at week 12, assuming that 12% of the patients in the placebo group would have an IGA response, and with at least 99% power to detect a between-group difference of 30 or more percentage points with respect to an EASI-75 response at week 12, assuming that 23% of the patients in the placebo group would have an EASI-75 response. We used a sequential, Bonferroni-based procedure to control the family-wise type I error rate at 5% for testing hypotheses across the two primary end points and the three key secondary end points. (Additional details are provided in Section S5 and Fig. S1 in the Supplementary Appendix.)
The two primary end points were tested first for the higher dose of abrocitinib and then for the lower dose at a significance level of 5%. To meet the primary objective, the differences between the abrocitinib dose groups and the placebo group with respect to the primary end points had to be statistically significant. With regard to the key secondary end points, the significance level was evenly split (2.5% each) for testing along two hierarchical sequences — sequence A was for the four comparisons of itch response at week 2 (200-mg abrocitinib vs. placebo, 100-mg abrocitinib vs. placebo, 200-mg abrocitinib vs. dupilumab, and 100-mg abroctinib vs. dupilumab), and sequence B was for the four comparisons of an IGA response (200-mg abrocitinib vs. placebo, and 100-mg abrocitinib vs. placebo) and an EASI-75 response (200-mg abrocitinib vs. placebo, and 100-mg abrocitinib vs. placebo) at week 16 (Fig. 2 in the statistical analysis plan, available in the protocol). The analyses of the primary and key secondary end points were based on available data up to and including week 16, with overall family-wise type I error controlled as specified in the statistical analysis plan. If a patient withdrew from the trial and no drug or placebo was dispensed at week 16, all available data for that patient was included in the week-16 analysis. There was no plan for adjustment of confidence intervals of the secondary end points after the key secondary end points, and no definite conclusions can be drawn from these data.
The primary analysis of efficacy was performed in the modified intention-to treat-population, which included all the patients who had undergone randomization and received at least 1 dose of a trial drug or placebo. Primary, key secondary, and all other binary end points were analyzed with the use of two approaches. In the first approach, the Cochran–Mantel–Haenszel statistic, with adjustment for baseline disease severity (moderate or severe), was used to test the hypothesis that there would be no differences between the two trial groups with respect to the primary and key secondary end points; the P values from the Cochran–Mantel–Haenszel test were used to test the hypothesis that there would be no differences between the two trial groups that were compared with respect to the other binary end points. In the second approach, the percentages of patients in each trial group who had responses were reported, and the differences between the two trial groups that were compared were summarized as weighted differences (based on the Cochran–Mantel–Haenszel statistic) and 95% confidence intervals, calculated with the use of normal approximation.
In the analysis of the end points beyond the three key secondary end points, there was also no plan for the adjustment of confidence intervals for multiple comparisons, and no inferences can be drawn from these data. Sensitivity analyses of the two primary end points are described in Section S6.
Continuous end points were analyzed with a mixed-effect model with repeated measures that used all observed data; the model included trial group, baseline severity, visit, trial-group–by–visit interaction, and relevant baseline values as fixed effects. In the analysis of continuous end points, no imputations were made for missing data because the mixed-effect model with repeated measures yielded valid inferences under the assumption of a missing-at-random mechanism. In the analysis of the binary end points, missing responses for the patients who had permanently withdrawn from the trial were defined as no response with respect to the primary end points at all visits after withdrawal; any observations that were missing intermittently (including baseline values) were considered to be missing completely at random and remained as missing in the analysis. Additional details of the analyses are provided in the statistical analysis plan.




留言 (0)