Patients
Eligible patients were 18 years of age or older, had received a diagnosis of psoriatic arthritis, fulfilled the Classification Criteria for Psoriatic Arthritis,6 and had historical or current plaque psoriasis. All the patients had at least 3 swollen joints (of 66 tested) and at least 3 tender joints (of 68 tested) at baseline, the presence at screening of one or more erosions on the hands or feet on radiography (as determined by central imaging review) or a high-sensitivity C-reactive protein level that was greater than the laboratory-defined upper limit of the normal range, and an inadequate response or unacceptable side effects with at least one nonbiologic DMARD. Stable treatment with nonsteroidal antiinflammatory drugs (NSAIDs), glucocorticoids, and no more than two nonbiologic DMARDs was permitted but not required. Patients were excluded if they had previous exposure to biologic therapies or JAK inhibitors. Details regarding inclusion and exclusion criteria are provided in Section S2 in the Supplementary Appendix, available with the full text of this article at NEJM.org.
Trial Design
The trial was conducted at 281 sites in 45 countries and was initiated in April 2017; the last patient completed week 24 in December 2019 (Table S1 in the Supplementary Appendix). Results for efficacy and safety are presented through week 24 (with the primary end point at week 12). The extension period is ongoing, with up to 3 years of total anticipated trial participation (Fig. S1).
Patients were randomly assigned by means of an interactive-response system in a 1:1:1:1 ratio to receive oral upadacitinib at a dose of either 15 mg or 30 mg once daily, placebo followed by upadacitinib at a dose of 15 mg or 30 mg once daily (1:1 ratio) starting at week 24, or subcutaneous adalimumab at a dose of 40 mg every other week. Randomization was stratified according to the extent of psoriasis (≥3% vs. <3% of body-surface area), current use or nonuse of at least one nonbiologic DMARD, the presence or absence of dactylitis, and the presence or absence of enthesitis.
Starting at week 16, patients who did not have at least 20% improvement in tender and swollen joint counts as compared with baseline at weeks 12 and 16 could initiate background treatment with DMARDs, NSAIDs, acetaminophen, low-potency opioids, or glucocorticoids or adjust the dose if they were already receiving the drug. During the 24-week placebo-controlled period, investigators, patients, and the sponsor were unaware of the trial group assignments.
Trial Oversight
The trial was conducted according to the International Council for Harmonisation guidelines and the principles of the Declaration of Helsinki. All the patients provided written informed consent. The trial protocol, available at NEJM.org, was approved by an independent ethics committee or institutional review board at each site.
The trial was sponsored by AbbVie, which provided upadacitinib, adalimumab, and placebo. Representatives of AbbVie designed the trial, participated in the collection and interpretation of the data, and paid for professional writing assistance, in addition to managing the data collection, maintaining the trial database, and performing the statistical analysis. Confidentiality agreements were in place between the authors and AbbVie. Data were collected by the investigators, their teams, and AbbVie. All the authors contributed to the development of the manuscript with the assistance of a professional medical writer, including interpretation of the data, and approved the final draft for submission. The authors vouch for the completeness and accuracy of the data, for the fidelity of the trial to the protocol, and for the accurate reporting of adverse events.
End Points
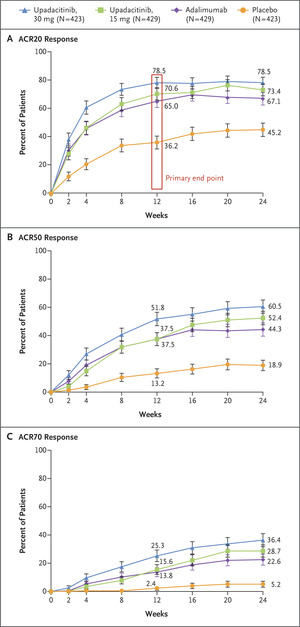
The primary end point was at least 20% improvement according to the ACR criteria (ACR20 response) with upadacitinib as compared with placebo at week 12. This end point represents a decrease from baseline of at least 20% in the number of tender and swollen joints and an improvement of at least 20% in at least three of five other domains (an assessment of disease activity on a numerical rating scale by both the patient and the physician, an assessment of disability level based on a patient questionnaire, the patient’s assessment of pain on a numerical rating scale, and high-sensitivity C-reactive protein level).
There were 14 multiplicity-controlled secondary end points: the change from baseline in the Health Assessment Questionnaire–Disability Index (HAQ-DI) score (ranging from 0 to 3, with higher scores indicating greater disability) at week 127; the percentage of patients with a score of 0 or 1 and at least a 2-point decrease from baseline on the Static Investigator Global Assessment (sIGA) of Psoriasis (ranging from 0 to 4, with higher scores indicating more severe skin involvement) at week 16,8 which was assessed in patients who had a baseline score of at least 2; the percentage of patients with a decrease from baseline of at least 75% in the score on the Psoriasis Area and Severity Index (PASI; ranging from 0 to 72, with higher scores indicating more severe disease) (PASI75 response) at week 16,9 which was assessed in patients who had an affected body-surface of at least 3% at baseline; the change from baseline in the modified total Sharp–van der Heijde Score (ranging from 0 to 528, with higher scores indicating greater damage) at week 24; the percentage of patients with minimal disease activity (determined by fulfilling five of seven criteria: a tender-joint count of ≤1; a swollen-joint count of ≤1; a PASI score of ≤1 or an affected body-surface area of ≤3%; a score on the patient’s assessment of pain of ≤1.5 [ranging from 0 to 10, with higher scores indicating more pain]; a score on the patient’s global assessment of disease activity of ≤2 [ranging from 0 to 10, with higher scores indicating more disease activity]; a HAQ-DI score of ≤0.5; and a score on the Leeds Enthesitis Index [LEI] of ≤1 [ranging from 0 to 6, with higher scores indicating more affected sites]) at week 2410; the percentage of patients with resolution of enthesitis (LEI score, 0) at week 24,11 which was assessed in patients with a baseline LEI score greater than 0; noninferiority of upadacitinib to adalimumab for the ACR20 response at week 12; the change from baseline in the score on the 36-Item Short Form Health Survey Physical Component Summary (SF-36 PCS; norm-based scores were used, with higher scores indicating better health-related quality of life) at week 1212; the change from baseline in the score on the Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-Fatigue; ranging from 0 to 52, with higher scores indicating less fatigue) at week 1213; superiority of upadacitinib to adalimumab for the ACR20 response at week 12; the percentage of patients with resolution of dactylitis (score of 0 on the Leeds Dactylitis Index [LDI; higher scores indicate more affected sites]) at week 24,14 which was assessed in patients with a baseline LDI score greater than 0; superiority of upadacitinib to adalimumab for the change from baseline in the patient’s assessment of pain (see above) at week 12; superiority of upadacitinib to adalimumab for the change from baseline in the HAQ-DI score at week 12; and the change from baseline in the score on the Self-Assessment of Psoriasis Symptoms (ranging from 0 to 110, with higher scores indicating greater severity of symptoms) at week 16.15 Additional end points, which were not adjusted for multiplicity, are shown in Table S2. The comparisons of upadacitinib with placebo and with adalimumab that are included here were prespecified.
Changes were made to the protocol and statistical analysis plan during the conduct of the trial to address regulatory-agency feedback regarding the method for noninferiority testing for upadacitinib as compared with adalimumab and for ordering of multiplicity-controlled end points on the basis of data from other trials. All changes were made before the database lock and unmasking of data. The primary and secondary end points were not altered, with the exception of revision of change from baseline in enthesitis to resolution of enthesitis and change from baseline in dactylitis to resolution of dactylitis, removal of the Hochberg test to adjust for multiple comparisons, and revision of the ordering of change in the FACIT-Fatigue score and resolution of dactylitis. Amendments are provided in Section S3 in the Supplementary Appendix.
Safety
Adverse event reporting and clinical laboratory testing were performed by investigators who were unaware of the trial group assignments, and safety results are reported through week 24. An independent, external cardiovascular adjudication committee, whose members were unaware of the trial group assignments, adjudicated deaths and cardiovascular events using prespecified definitions.
Statistical Analysis
Efficacy analyses were conducted in the modified intention-to-treat population, which included all the patients who had undergone randomization and had received at least one dose of upadacitinib, placebo, or adalimumab. A sample size of 1640 patients was planned to provide at least 90% power to detect a difference between upadacitinib and placebo for the primary end point and for most key secondary end points and at least 85% power for evaluating the noninferiority and superiority of each upadacitinib dose as compared with adalimumab with respect to the ACR20 response at week 12. The trial was not powered to compare upadacitinib with adalimumab regarding the change from baseline in pain and in the HAQ-DI score. All power and sample-size calculations were performed at a two-sided significance level of 0.025, with a dropout rate of 10% taken into account.
The overall type I error rate for the calculations of the primary end point and the 14 ranked secondary end points was controlled with the use of a two-part sequential graphical multiple-testing procedure. Part 1 started with the primary end point, with the use of α÷2 (α was 0.0499) for each dose followed by a prespecified hierarchical α transfer path that included downstream transfer along the end-point sequence within each dose as well as cross-dose transfer between each upadacitinib dose (Fig. S2). Part 2 was tested only if the results for all end points for both doses in Part 1 were significant. If the results for some end points in Part 1 were not significant, then no α was passed to part 2 of the analysis. The end points in part 2 were tested with the use of level α in a fixed sequence. Once the results for an end point were deemed to be significant, its significance level was transferred to subsequent end points following the prespecified order. When the hierarchical analysis failed, subsequent end points were not tested and only point estimates with multiplicity-unadjusted 95% confidence intervals are given.
For binary end points, trial groups were compared with the use of the Cochran–Mantel–Haenszel test, with adjustment for current DMARD use for each upadacitinib dose as compared with placebo. Imputation of nonresponse was used for the handling of missing data. Between-group differences in the percentage of patients with a response and the associated 95% confidence intervals with the use of normal approximation for the between-group difference are presented. For the percentage of patients with an ACR20 response at week 12, the noninferiority of each upadacitinib dose to adalimumab was assessed with the use of Koch’s three-group approach; noninferiority was achieved if upadacitinib preserved at least 50% of the placebo-subtracted adalimumab effect. Additional details are provided in Section S4 in the Supplementary Appendix. The original plan for noninferiority comparison of upadacitinib with adalimumab for the ACR20 response at week 12 used a margin of 15 percentage points, and this analysis is also presented.
For nonradiographic continuous end points, analyses were conducted with the use of a mixed-effects model for repeated measures (MMRM) with fixed effects of treatment, visit, treatment-by-visit interaction, current DMARD use (yes vs. no), and the corresponding baseline value as a covariate. Missing data were handled by means of MMRM, with the assumption of missing data at random. Least-squares mean differences and the associated 95% confidence intervals are provided with the use of MMRM. Prespecified sensitivity analyses that used the tipping-point method were performed for the change from baseline in the HAQ-DI score. Post hoc sensitivity analyses were performed for the SF-36 PCS and FACIT-Fatigue scores (Table S3). The confidence intervals for differences between groups for additional secondary end points and sensitivity analyses were not adjusted for multiplicity, and no clinical inferences can be drawn from those data. Additional details are provided in the statistical analysis plan, available with the protocol at NEJM.org.




留言 (0)