
記住我




Templated reactions provide a powerful toolbox for the conditional control of chemical reactions and molecule activity. Utilizing the unique Watson-Crick-based recognition properties of short oligonucleotide probes, reactive groups of attached payloads can be arranged in spatial proximity, allowing a reaction to occur efficiently at low concentrations of reactants when the rate of non-templated bimolecular reactions is extremely low. A vast number of different nucleic acid-encoded chemistries are available today, comprising ligations, bond cleavage reactions and functional group interconversions.[1, 2] Although many research groups have designed templated reaction systems for a DNA- or RNA-controlled activation of fluorescent reporter dyes, much fewer examples have been reported that utilize oligonucleotide-templated reactions as a means to control the bioactivity of drug-like molecules.[3] This is due to a critical design constraint of such systems: the reaction must be able to create a pharmacophore, ideally from a completely inactive precursor. This is difficult to achieve in a single reaction step. Most templated drug-release systems have therefore relied on dissociative chemistries that produce a bioactive molecule by bond cleavage, such as ester hydrolysis.[4-9] A disadvantage of these systems is that they are prone to premature fragmentation and not all drug candidates can be effectively inactivated by attachment to a cleavable linker. Few examples have demonstrated that activity can instead also be created from bond formation, such as acyl transfer.[10-13]
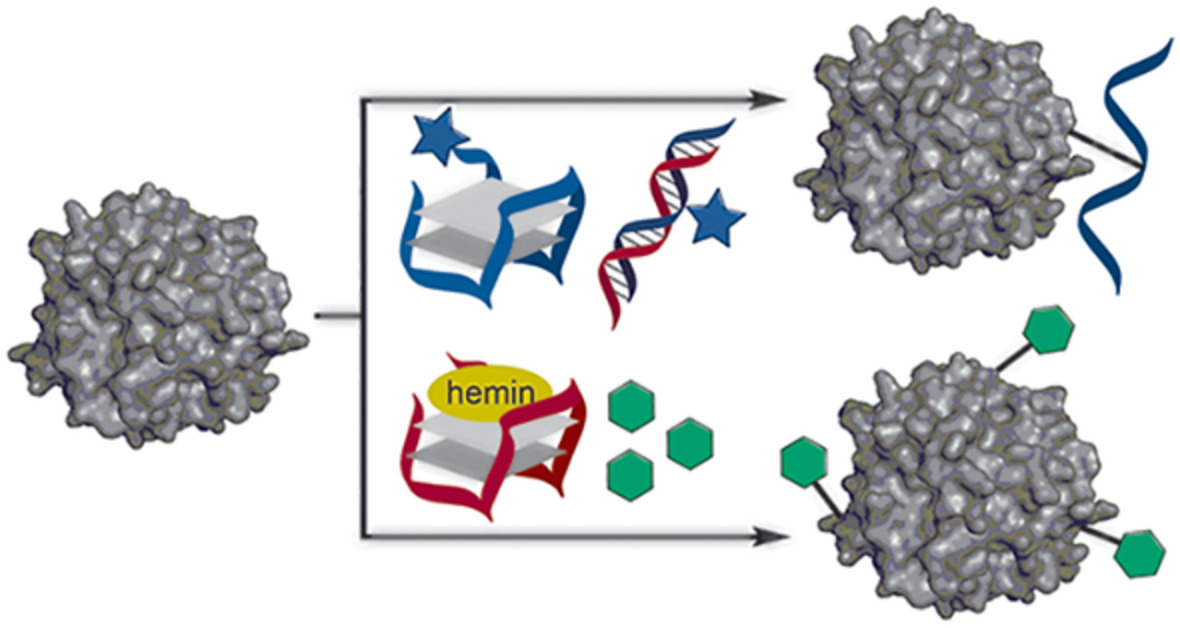
Nucleic acid templated chemistries offer the possibility of turnover.[14-16] Reaction systems that allow a single template molecule to instruct the formation of several product molecules have been developed with the aim to increase the sensitivity of nucleic acid detection. Such amplification systems could also be of interest for approaches in which nucleic acid templates control the bioactivity of a payload attached to nucleic acid molecules. In an effort to explore new options for templated drug activation, we considered Smac mimetic compounds (SMCs). These proapoptotic drugs are modeled after the N-terminal AVPI tetrapeptide motif of Smac, the natural antagonist of the Inhibitor of Apoptosis Protein (IAP) family, which is overexpressed by many human cancers.[17, 18] Like their parent, SMCs bind to IAP baculoviral IAP repeat (BIR) domains to prevent interaction with target caspases and cause rapid autoubiquitylation and proteasomal degradation. X-ray and NMR-based structure-activity relationship studies revealed a central importance of their N-terminal amino group for SMC affinity: It is involved in multiple, essential hydrogen bridges with Asp-214, Glu residues 219 and 324 and Gln-319 residues of a small binding pocket in both BIR2 and BIR3 (Figure 1A).[19-21] Mono N-methylation, which is often found in SMCs to enhance pharmacokinetic properties, is tolerated, but N,N-dimethylation, acylation or substitution with a methyl group increases the Kd by several orders of magnitude, rendering the molecule inactive.[19, 22]

A, Structure of Smac mimetic MV1 and the importance of its N-terminal amine for X-linked Inhibitor of Apoptosis Protein (XIAP) BIR2/BIR3 domain binding. B, Outline of an XIAP mRNA-templated system for a reductive activation of azide-modified Smac mimetics compounds (SMC) with phosphines or ruthenium complexes
We speculated that this property could be exploited to put SMC activity under the control of an oligonucleotide-templated reaction. We envisioned that the introduction of an azide will enable a conditional “on switching” of SMC activity (Figure 1B) by reduction to the amine. Azides accept but cannot donate hydrogen bonds and should, therefore, not be able to interact with the critical residues in the binding pocket. Moreover, azides are stable to conditions typically found in biological systems, offering prospects for a sequence-dependent “uncaging” of SMC bioactivity: Controlled by a conjugated oligonucleotide, the activating azide reduction would only occur in cells that express significant amounts of a specific template sequence. This could be used to directly counteract the effects of IAP overexpression by utilizing their mRNA to produce the inhibitor.
Herein we report the RNA-templated conversion of azido-functionalized peptides to Smac mimetic molecules. We compared two reactions, the templated Staudinger reaction with phosphines and Ru2+ complex-catalyzed photoreduction. Both reactions have previously been successfully applied in templated chemistries, to restore fluorescence of quenched fluorophores or to release bioactive molecules such as isopropyl-β-d-thiogalactopyranoside (IPTG) or estradiol via cleavage of p-azido(methyl)benzyloxycarbonyl linkers.[23-27] In contrast, we explore templated azide reduction as a way to unleash a peptide's N-terminal amino group without involving linkers. For recognition of the RNA template, the azidopeptides were appended to peptide nucleic acids (PNA) and 2′-OMe oligoribonucleotides. The comparison of both recognition units showed surprising differences in reactivity when the reactive probes were applied in excess to the RNA template.
2 MATERIALS AND METHODSBuilding blocks, resins and coupling agents were obtained from the commercial suppliers stated in the procedures or listed in the supporting information. Prior to coupling, l-2-azido propionic acid CHA salt (ChemPep, Wellington, Florida) was converted into its acid form by extraction from dichloromethane (0.2 M) and hydrochloric acid (0.5 M). HPLC-purified RNA templates and amino-modified 2′-OMe-RNA oligonucleotides were purchased from biomers.net (Ulm, Germany). Tris(2-carboxyethyl)phosphine dimethyl ester (dmTCEP) was prepared according to a literature procedure.[28]
2.1 Protein expression and purificationX-linked IAP (XIAP) L-BIR2-BIR3 protein (residues 120-356) containing an N-terminal 6x His-tag was expressed in Escherichia coli BL21(DE3) grown at 37 °C in LB medium supplemented with Kanamycin (50 μg/mL). Upon reaching an OD600 of 0.6, protein expression was induced by addition of 0.4 mM IPTG and 100 μM ZnAc2 for 20 hours at 20 °C. Cells were lysed in buffer (50 mM Tris, 200 mM NaCl, 50 μM ZnAc2, 0.1% β-mercaptoethanol, pH = 7.5) containing 1 cOmplete Mini protease inhibitor tablet (Sigma-Aldrich, St. Louis, Missouri) per 10 mL, using a French press. Lysates were centrifuged (45 000 rcf, 30 minutes, 4 °C) and the soluble fraction was purified using a 5 mL HisTrapHP Ni column (25, 62.5, 200, 350, 500 mM imidazole) followed by gel filtration on a Superdex 75 column in buffer (20 mM Tris, 200 mM NaCl, 50 μM ZnAc2, 1 mM DTT, pH 7.5). After purification, glycerol and DTT were added to a final concentration of 10% (vol/vol) and 10 mM.
2.2 Fluorescence polarization binding assayRelative binding affinity (IC50) was measured as described by Nikolovska et al.[29] Fluorescein-labeled reference binder Smac-1F was synthesized according to the procedure described in the literature. To each well of a black, nonbinding 96-well plate, 1 nM of Smac-1F and 15 nM l-BIR2-BIR3 construct in assay buffer (100 mM KH2PO4/K2HPO4, 110 mM NaCl, 5 mM DTT, 100 μg/mL bovine γ-globulin, 0.02% NaN3) was added and mixed with different concentrations of Smac mimetics. The plate was incubated for 2 hours in the dark at room temperature and the anisotropy values were determined using a plate reader (Ex./Em. λ = 485/535 nm).
2.3 Synthesis of peptides and PNA conjugatesSynthesis of peptides and PNA conjugates was conducted by automated Fmoc SPPS on ChemMatrix Rink Amide resin (peptides; 35-100 mesh, 0.5-0.7 mmol/g; Sigma-Aldrich), TentaGel XV Rink Amide (conjugates; 100-200 mesh; 0.23 mmol/g) or TentaGel S Trt Cl resin (peptide acids; 90 μm; 0.2-0.3 mmol/g; Rapp-Polymere, Tübingen, Germany) using a ResPep SL parallel peptide synthesizer (Intavis Bioanalytical Instruments, Cologne, Germany). The Fmoc group was removed with 20% piperidine/DMF. Fmoc/Bhoc protected PNA monomers (4 eq.) were coupled with HCTU/NMM, Fmoc-protected amino acids (6 eq.) with HCTU/OxymaPure/NMM. Couplings were performed for 30 minutes (60 minutes for Fmoc-Chg-OH) at room temperature with 2 minutes of preactivation and repeated once. Acetylation of the N-terminus/capping of unreacted building blocks was performed by treatment with 5% Ac2O, 6% 2,6-lutidine in DMF for 2 minutes (2x). Reducing agents were coupled to the side chain amine of 1Lys or 1Dap after removal of the Alloc protecting group with Pd(PPh3)4 (0.1 eq.) and PhSiH (20 eq) in degassed, dry dichloromethane (20 minutes, 2x). dmTCEP (6 eq.) was coupled with DIC/OxymaPure in degassed DMF for 1 hour, Ru(bpy)2(mcbpy) (1.1 eq.) was introduced as NHS ester in the presence of DIPEA for 24 hours. Sequences were cleaved off the solid support with 96% TFA, 2% TIS, 2% H2O for 2 hours, precipitated in ice-cold Et2O and/or dried under an argon stream. Crude peptides were purified by RP-HPLC on a Nucleodur Gravity C18 column (250/10 mm, 5 μm, 110 Å; Macherey-Nagel, Düren, Germany) with a gradient of eluent A1 (1% MeCN /0.1% TFA in water) and eluent B1 (1% water/0.1% TFA in MeCN). Detailed experimental procedures, yields and characterization data are provided in the supporting information.
2.4 Synthesis of 2′-OMe-RNA conjugatesN3-SMC-2′-OMe-RNA conjugates were prepared by an in-solution coupling procedure: 3′-C6-amino-linker-modified 2′-OMe-RNA (1 eq.) and DIPEA (10 eq.) were dissolved in 10% water/DMSO at a concentration of 2 μM. N3-SMC acid (20 eq.), HATU (19 eq) and DIPEA (20 eq.) were dissolved in DMSO (final concentration N3-SMC: 0.2 M), preactivated for 2 minutes and added to the 2′-OMe-RNA solution. The mixture was shaken for 4 hours at RT. Ru(bpy)2(mcbpy) was introduced as NHS ester: 5′-C6-amino-linker-modified 2′-OMe-RNA (1 eq.) was dissolved in borate buffer (100 mM Na2B4O7, pH 8.5) at a concentration of 500 μM. Ru(bpy)2(mcbpy-O-Su-ester)(PF6)2 (3 eq.) in DMSO (15 mM) was added and the solution was shaken at RT for 2 hours. The products of both reactions were isolated by semipreparative HPLC on a Triart C18 column (150/10 mm, 5 μm, 120 Å; YMC Europe, Dinslaken, Germany) with a gradient of eluent A2 (0.1 M TEAA in water) and eluent B2 (MeCN).
2.5 Templated reactionsTemplated reactions were performed in degassed MOPS buffer (10 mM MOPS, 200 mM NaCl, 0.1% [wt/vol] CHAPS, pH 7.0) using UPLC vials with poplypropylene microvolume inserts (dmTCEP system; Sigma-Aldrich) or black, nonbinding 96-well F-bottom plates (Ru(II) system; Greiner Bio-One, Kremsmünster, Austria) as reaction vessels. For templated photoreductions, the buffer was supplemented with 5 mM sodium ascorbate. A reaction temperature of 25 °C or 37 °C was maintained using the sample manager compartment of an Acquity UPLC system (dmTCEP system; Waters, Milford, Massachusetts) or a custom-built multiwell plate heating element connected to a thermostat (Ru(II) system). Mono- or bivalent N3-SMC-probe (1 μM) and dmTCEP/Ru(bpy)2(mcbpy)-PNA (2 or 3 μM) were mixed in pretempered buffer and allowed to react for up to 360 minutes in the presence or absence of stoichiometric/substoichiometric amounts of RNA template (1, 0.3, 0.1, 0.03, 0.01, 0 eq.). Ru(II) system samples were irradiated (1 W, λ = 455 nm, current limit 1000 mA, intensity 98%) with a collimated LED lamp (M455L3-C1, Thorlabs, Newton, New Jersey) mounted 15 cm above and controlled by a DC2200 LED driver (Thorlabs). At different timepoints, samples were withdrawn (dmTCEP: automatically, Ru(II): manually) and analyzed on an Acquity UPLC system (PNA probes: Acquity UPLC CSH C18 column; 2.1 × 100 mm, 1.7 μm, 130 Å; 2′-OMe-RNA probes: Acquity UPLC Oligonucleotide BEH C18 column, 2.1 × 50 mm; 1.7 μm; 130 Å; Waters) using optimized linear gradients (PNA probes: 10%-80% B in 3 minutes; eluent A = 1% MeCN /0.1% TFA in water, eluent B = 1% water/0.1% TFA in MeCN; 2′-OMe probes: 3%-70% B in 3 minutes; eluent A = 0.1 M TEAA in water, eluent B = MeCN). The probe conversion was calculated from the area under the curve of the educt ( ) and product (
) and product ( peaks as:
peaks as:  . Data were collected from at least two independent experiments.
. Data were collected from at least two independent experiments.
Melting curves were measured on a V-750 spectrophotometer equipped with a PAC-743R Peltier cell changer (JASCO, Tokyo, Japan) and connected to a F250 recirculating cooler (JULABO, Seelbach, Germany). Solutions containing PNA conjugate or 2′-OMe-RNA (1 μM) and a complementary RNA (1 μM) were prepared in phosphate buffer (10 mM NaH2PO4/Na2HPO4, 100 mM NaCl, pH = 7.0). The duplex absorption (λ = 260 nm) was determined between 18 °C and 90 °C (0.5 °C/min) at a sampling rate of 5 points/°C. Three measurements were averaged and the Tm was determined as maximum of the first derivative of a sigmoidal fit.
3 RESULTS AND DISCUSSION 3.1 Binding affinity of N-terminally modified SMCsIn a preliminary experiment, we tested whether substituting the N-terminal amine of Smac mimetic peptides with an azide achieves a similar reduction of binding affinity to XIAP as acetylation. Since azide reduction can only produce a primary amine, not N-methylamine, like it is found in many SMCs, this experiment should also shed light on potential affinity differences between those two modifications. For the test, two tetrapeptide sequences were selected: the original N-terminal Smac motif Ala-Val-Pro-Ile and the unnatural variant MeAla-Chg-Pro-Dip, also known as MV1.[30] Of each, four derivatives were synthesized, bearing either Ala, N-methyl-Ala (MeAla), 2-azidopropionic acid (N3Ala) or N-acetyl-Ala (AcAla) in the P1 position.
The modified peptides were then tested in a fluorescence polarization-based competitive binding assay with a recombinantly expressed XIAP protein, comprising the BIR2 and BIR3 domains relevant for SMC binding (residues 120-356) and a fluorescent reference binder Smac-1F.[29] Gratifyingly, the H2N- and MeHN-modified sequences had similar affinities (Table 1, see also Figure S1), indicating that the product of a templated reaction would not suffer from reduced affinity. Unmodified peptide 1 bound the XIAP protein with an IC50 of 3.28 μM, while peptidomimetic binder 5 demonstrated slightly higher affinity with 0.48 μM. The MeHN-modified sequences 2 and 6 were only marginally different (3.63 and 1.12 μM). The higher affinity of the MV1-based sequence is likely the result of the optimized residues in P2 and P4. As expected, acetylation reduced binding affinity by four orders of magnitude (IC50 (4) ≥ 10 mM, IC50 (8) = 2.0 mM). Replacing the amine with an azide was only slightly less inactivating. Azido-SMC 3 showed an IC50 value of 1.5 mM, while 7 had a half-maximal affinity of 0.6 mM. This confirmed that hydrogen bonding between the BIR domains and the N-terminal azide is not taking place. Conceivably, the larger size of N3 vs NH2 may also contribute to the inactivating effect, as the P1 binding pocket is relatively small. An additional experiment, in which 3 was treated with 10 eq. TCEP for 1 hour prior to the binding assay, verified that azido-SMC affinity to XIAP can be fully restored by Staudinger reduction (Figure S2).
TABLE 1. Binding affinity (IC50) of N-terminally modified Smac mimetic peptides to a recombinant XIAP-L-BIR2-BIR3 protein Peptide R = Nr IC50 (M) R-Ala-Val-Pro-Ile-NH2 H2N 1 3.28 ± 0.90 × 10−6 MeHN 2 3.63 ± 1.39 × 10−6 N3 3 1.50 ± 0.82 × 10−3 AcHN 4 ≥1.00 × 10−2 R-Ala-Chg-Pro-Dip-NH2 H2N 5 0.49 ± 1.18 × 10−6 MeHN 6 1.12 ± 0.12 × 10−6 N3 7 5.61 ± 0.34 × 10−4 AcHN 8 1.98 ± 0.50 × 10−3 Note: Determined in a competitive FP assay with 2 nM reference binder Smac-1F and 20 nM XIAP-L-BIR2-BIR3 in buffer (100 mM KH2PO4/K2HPO4, 100 μg/mL bovine γ-globulin, 0.02% NaN3, pH 7.5). Abbreviation: XIAP, X-linked Inhibitor of Apoptosis Protein.It should be noted that the binding affinity of a PNA- or 2′-OMe-RNA-appended SMC may be different to that of the free inhibitor, due to the steric bulk added by the oligomer. However, previous publications have demonstrated that such conjugates are indeed active and able to activate caspases in cell lysate[10, 11] or induce apoptosis in human cancer cell lines.[31, 32]
3.2 Templated Staudinger reductionNext, we prepared a set of PNA conjugates to test the reductive activation in a templated reaction (Figure 2). PNA was used due to its high binding affinity, excellent sequence specificity and compatibility with solid phase peptide synthesis.[33, 34] The probes were designed to be complementary to a stretch of mRNA coding for the X-linked Inhibitor of Apoptosis Protein (XIAP). This potent member of the IAP family mediates apoptosis resistance in many human cancers by directly inhibiting caspases 3, 7 and 9.[35] To increase solubility of the relatively hydrophobic conjugates, Lys and Glu residues were added to the N- or C-terminal end of the PNA, respectively. The combination of a basic and an acidic amino acid creates a net neutral zwitterion, which should not induce non-specific electrostatic interactions with the RNA template, but still mitigate PNA aggregation by providing additional options for solvation.

RNA-templated reduction of mono- or bivalent azido-Smac mimetic compound (SMC)-probes with tris(2-carboxyethyl)phosphine dimethyl ester (dmTCEP)- or Ru(bpy)2(mcbpy)-peptide nucleic acids (PNA). Chg = l-cyclohexylglycine, Dip = l-diphenylalanine, N3Ala = l-2-azido propionic acid. Position numbering of probes and template is relative to the start codon of X-linked Inhibitor of Apoptosis Protein (XIAP) mRNA (NCBI accession number: NM_001167.4)
For the azido-SMC-bearing probes, the MV1-based sequence was used. It binds more strongly to XIAP and is potentially more metabolically stable due to the presence of unnatural amino acids. We explored both monovalent (N3) and bivalent ((N3)2) azido-SMC-PNA conjugates. Dimerized Smac mimetics possess significantly higher affinity for IAPs than their monovalent counterparts, as they can concurrently interact with two BIR domains, mimicking the mode of action of their homodimeric parent Smac. The reduced form of (N3)2 mimics the structure of BV6, a bivalent version of MV1.[30] As reducing probe, we tested dmTCEP, a more reactive and cell-permeable derivative of TCEP,[36] and the ruthenium complex Ru(bpy)2(mcbpy), both conjugated with PNA.[37] The latter acts as photoredox catalyst when excited by blue light (λmax = 452 nm), which adds an extra element of temporal control to the templated reaction that may prove useful in a further application of the reaction system in a biological context. Ru(bpy)2(mcbpy) also requires stoichiometric amounts of a sacrificial electron donor such as sodium ascorbate (NaAsc) or NADPH to be reactive. Winssinger and coworkers recently demonstrated that the cellular redox pool provides high enough concentrations of these molecules to allow Ru(II)-catalyzed reactions to proceed within living cells.[27, 38]
To reduce oxidation to the phosphine oxide, dmTCEP was coupled to the ε-amino group of 1Lys under base-free conditions using DIC/Oxyma, while the ruthenium complex was introduced as NHS ester. To assess the role of proximity between reducing agent and azide, PNA probes were complementary to three different positions on the template, creating a gap of one to three unpaired nucleotides.
In the phosphine-based templated Staudinger reduction system, a twofold excess of reducing agent probe was used to allow full conversion of both monovalent and the bivalent azidopeptide conjugates N3 and (N3)2, respectively. On equimolar amounts of XIAP template, reduction of monovalent N3-SMC-PNA N3 was fast, yielding more than 50% product within 5 minutes regardless of the gap size (Figure 3A and Table S1). Importantly, in the absence of RNA template only about 5% product was obtained after 6 hours. Comparing the maximum reaction speed vmax, the RNA template accelerated the reduction of N3 by more than 700-fold. Two observations are noteworthy. First, after a fast, initial phase, the reaction kinetics slowed down. This may be due to the increased number of products poisoning the templated reaction by increasing the probability of unproductive alignments after strand exchange reactions. Second, the templated reaction did not reach full conversion and leveled at 70% to 80% yield. This is due to the emergence of two side-products (Figure S3). Their amounts were too low to allow identification. We noticed, however, that the side reaction only occurred when dmTCEP-PNA and N3-SMC-PNA were allowed to react in presence of the template. No side reaction occurred when N3-SMC-PNA was incubated with triscarboxyethylphosphine (data not shown). Perhaps, the aza-ylide intermediate formed upon Staudinger reaction of dmTCEP facilitates hydrolysis of a methyl ester unit.

Templated Staudinger reduction of 1 μM, A, mono- or, B, bivalent N3-Smac mimetic compound (SMC)-peptide nucleic acids (PNA)- with 2 or 3 μM dmTCEP-PNA 9-11. Reactions were performed at 25 °C in buffer (10 mM MOPS, 200 mM NaCl, 0.1% CHAPS, pH 7.0) in the presence or absence (no template [NT]) of 1 μM RNA template
TABLE 2. Tm of PNA and 2′-OMe-RNA sequences Probe Sequence Tm (°C) PNA XIAP211-219 ccatctatc 49.8 XIAP221-229 ctccatatt 46.2 2′-OMe-RNA XIAP211-219 CCAUCUAUC 39.1 XIAP208-219 CCAUCUAUCUAC 50.1 XIAP221-229 CUCCAUAUU 33.7 XIAP221-232 AGUCUCCAUAUU 45.0 Note: Measured at 1 μM in buffer (10 mM NaH2PO4/Na2HPO4, 100 mM NaCl, pH 7.0) with 1 μM complementary RNA. Abbreviations: XIAP, X-linked Inhibitor of Apoptosis Protein; PNA, peptide nucleic acids.Comparing the different dmTCEP probes, the lowest total yield was obtained with the combination of 11 and N3. Although the larger gap size between the annealing sites of these probes on the template is an obvious explanation for this, it should be noted that the PNA:RNA duplex of 11 also possesses a higher Tm (55.1 °C) than 9 (46.2 °C; Table 2, Figure S7). This can affect strand exchange reactions, as will be discussed later.
Next, we tested templated reactions of bivalent azidopeptide conjugate (N3)2. The templated reduction of (N3)2 with dmTCEP-PNA 9-11 will first lead to an intermediate in which only one of the two azide groups is reduced. The H2N-SMC-N3-conjugate rapidly formed at an initial rate (vmax = 2.72-3.03 nM∙s−1) comparable to reduction of the monovalent N3. Perhaps remarkably, the fully reduced (H2N-SMC)2 was observed already in the early reaction phase although the rate of formation was slower than that of N3 (vmax = 0.15-0.31 vs 1.80-1.98 nM∙s−1; Figure 3B and Table S1). This provides unambiguous evidence that strand replacement (of the dmTCEP-probes) occurs. For the combinations 9 and 10 + (N3)2, respectively, a plateau of about 60% conversion was reached after 360 minutes. The reason for the lower total yield compared to N3 was a combination of product inhibition exacerbated by the accumulating mono-reduced intermediate (H2N-R-N3), and additional formation of the unidentified byproducts in the second reduction step (Figure S3B). Interestingly, the slower reaction kinetics of the templated Staudinger reduction of bivalent (N3)2 increased the observable differences between the probes. The combination of 9 and (N3)2 (a gap of 1 nt) was significantly faster than the other two, reaching its half-maximal conversion after about 24 minutes (t1/2 = 74 minutes and 44 minutes for 10 and 11, respectively). The combination of 10 and (N3)2 eventually reached the same conversion level as 9 and (N3)2, whereas 11 and (N3)2 stagnated at about 40%. This is the same behavior as observed for the reduction of the monovalent conjugate and, as mentioned before, possibly related to the higher Tm of 11, which hinders strand exchange reactions required to bring in fresh reducing agent after the first reduction event.
From the experiments performed with phosphine probes, we infer that strand exchange reactions play multiple roles. While strand exchange obviously is required to improve the efficiency of reactions involving bivalent azido probes, it may also hamper progress at later phase of the reaction when product molecules compete with starting materials for binding of the template. Reactions in which a catalytically active moiety substitutes for the phosphine should not require displacement of the reductant strand enabling reactions to proceed rapidly also at later phases.
3.3 Templated photoreductionRuthenium complexes such as Ru(bpy)2(mcbpy) (Figure 1B) are suitable catalysts of azide reduction. As the ruthenium complex requires excitation and the presence of an electron donor to be active, the setup of the templated reaction was slightly modified. Samples were irradiated with a collimated LED lamp (1 W, λ = 455 nm) and 5 mM sodium ascorbate was added to the reaction buffer.
As already reported by others,[16, 26, 38, 39] the Ru(II)-catalyzed reduction was faster than the Staudinger reduction (vmax = up to 4.44 nM∙s−1 vs 1.98 nM∙s−1) and, in contrast to the phosphine reactions, proceeded well also at later reaction phases until 90% conversion within 20 to 30 minutes (Figure 4A and Table S1). Conversely, in the absence of RNA template, only 2% to 3% product was formed after 90 minutes. A gap of a single unpaired nucleotide (12 + N3) produced the most efficient reaction (t1/2 = 4.9 minutes), resulting in a relative speed increase over background of close to 900-fold. The reason for the slightly longer half-lives of Ru(II)-catalyzed reactions compared to the dmTCEP system, was the occurrence of a short lag-phase of 1 to 2 minutes in the beginning of the reaction. This was possibly due to the relatively low intensity of the LED light source used and thus time required to build up the reductively active Ru(I) species. In a separate experiment, we also verified the specificity of the templated reaction. When a scrambled azido-SMC probe scrN3 was used instead of N3, the same yield as in the non-templated reaction was obtained (Figure S6A). This confirmed that the catalytic effect of the RNA on the reaction rate is sequence-dependent.

Templated photoreduction of 1 μM, A, mono- or bivalent, B, N3-Smac mimetic compound (SMC)-peptide nucleic acids (PNA)- with 2 μM Ru(II)-PNA 12-14. Reactions were performed at 25 °C in buffer (10 mM MOPS, 200 mM NaCl, 5 mM NaAsc, 0.1% CHAPS, pH 7.0) in the presence or absence (no template [NT]) of 1 μM RNA template and with irradiation (LED, 0.98 W, λ = 455 nm)
The greater speed of the Ru(bpy)2(mcbpy)-based system illustrates the impact of the reducing agent-PNA no longer requiring strand exchange. As the Ru(II) complex acts catalytically, a single, target bound probe molecule can reduce multiple azide groups. This was especially evident for the templated reaction with bivalent SMC (N3)2 (Figure 4B). Here the kinetics were essentially the same as for the monovalent conjugate N3, and for the combination of 12 and (N3)2, about 90% conversion were achieved within 20 to 30 minutes (t1/2 = 4.6 minutes). Much less of the mono-reduced intermediate accumulated and no side products were formed (Figure S5), thus a much higher yield could be achieved than with the dmTCEP-based probes.
3.4 Comparison of PNA and 2′-OMe-RNAPNA has frequently been used as a biostable recognition unit in nucleic acid-templated chemistry. However, the hydrophobicity of PNA conjugates can be problematic (Figure S4). Most nucleic acid tools are based on oligonucleotide scaffolds. Phosphorothioate structures and 2′-O-methoxy ribonucleotides are frequently introduced into oligonucleotide tools to prevent degradation by nucleases. In a few studiesaddressing inhibition or localization of miRNA or mRNA was PNA compared side-by-side with metabolically stable oligonucleotide tools.[40-44] A comparison of different backbones in templated chemistry was, to our best knowledge, lacking and we, therefore, set out to explore the RNA-triggered photocatalytic reduction of azidopeptides on 2′-O-methoxy oligoribonucleotides scaffolds.
To maintain comparability between PNA- and 2′-OMe-RNA probes in templated reactions, they should possess the same affinity to the target RNA. To that end, the duplex melting temperatures of different 2′-OMe-RNA sequences were evaluated (Table 2, see also Figure S9). An initial test with the same 9-mer sequences as used for the PNA, showed Tm values more than 10 °C lower (39.1 °C and 33.7 °C vs 49.8 °C and 46.2 °C).
By increasing the length to 12-mers, almost matching melting temperatures (50.1 °C and 45.0 °C) could be obtained.
The 2′-OMe- and PNA-based Ru(II) probes were compared in templated reactions at different stoichiometric ratios of RNA target (0.01-1.0 eq.). This should reveal potential catalytic differences as well as determine the maximum achievable turnover of each system. To facilitate turnover in template, the reactions were measured at 37 °C. When the reactions were performed at a template concentration that matched the concentration of the azidopeptide, both PNA and 2′-OMe probe performed identically, reaching their maximum conversion (about 90%) after 15 minutes (Figure 5). Interestingly, at 37 °C, the lag phase of the ruthenium complex was a bit shorter (t1/2 = 3 minutes; Figure S6B). Also, the acceleration of the reaction over background by the template increased slightly, to around 1100-fold. When the amount of template was decreased, catalytic differences between the PNA- and 2′-OMe-RNA-based probes became apparent (Figure 5, Figure S7). In the presence of 0.3 eq. template (300 nM), the PNA probes achieved nearly the same yield as with 1 eq. RNA (albeit slightly later; t1/2 = 7.1 minutes), while the maximum conversion for the 2′-OMe-RNA probes dropped to 56%. With as little as 0.01 eq. (10 nM) of template RNA, the PNA-system still reached 15% conversion. At this concentration, the templated reaction of 2′-OMe-RNA probes provided only slightly more product than the non-templated background reaction, yielding only about 5% product after 60 minutes.

Turnover experiments with 2′-OMe- and peptide nucleic acids (PNA)-based Ru(II)-probes. Reaction kinetics for the reduction of 1 μM N3-Smac mimetic compound (SMC)-PNA N3 or N3-SMC-2′-OMe-RNA N3-2′-OMe with 2 μM of the corresponding Ru(II)-conjugate 12 or Ru-2′-OMe. Reactions were performed at 37 °C in buffer (10 mM MOPS, 200 mM NaCl, 5 mM NaAsc, 0.1% CHAPS, pH 7.0) in the presence of varying amounts of RNA (0.01-1.0 μM) or no template (NT) and under irradiation (LED, 0.98 W, λ = 455 nm)
The experiments point to significant differences between RNA-templated photoreduction of azidopeptides on PNA and 2′-OMe-RNA probes. First, the yields obtained with PNA probes are higher. Second, the reaction with PNA is faster, especially when photoreduction occurs at low template to azidopeptide ratios. One factor that allows the PNA conjugates to achieve better conversion than the 2′-OMe congeners is most likely a different stability of the Ru(II)-complexes. The UPLC measurements revealed that significant amounts of catalyst degrade during irradiation, and this effect is more pronounced for the 2′-OMe-RNA-based probes (Figure S8A). PAGE analysis of irradiated and nonirradiated samples (Figure S8C) showed no fragmentation of the conjugate, so a photocatalytic cleavage of the phosphodiester bonds, as has been reported for other Ru complexes,[45, 46] can be ruled out. Instead it is likely an oxidative process (a mass difference of +16 could be detected for one of the degradation products; Figure S8B). Ruthenium polypyridyl complexes have been shown to cause oxidative damage to nucleic acids, particularly guanine,[47, 48] of which the longer 2′-OMe-RNA probe has one more than the PNA. It is also conceivable that the latter generally has lower susceptibility to oxidative damage, as PNA has been found able to suppress photooxidation of a hybridized DNA.[49] However, the differential stability of the Ru(II)-complexes cannot account for the differences in reaction rates at early phases because the amount of intact Ru(II)-strand remains identical within the first 10 minutes of the reaction (Figure S7A). While the initial rates on one equivalent template are similar, reactions on PNA occur at twice the speed when the photoreduction is performed under turnover conditions. This indicates a different strand exchange behavior. To achieve a similar Tm, 2′-OMe-RNA probes are longer than PNA probes. The dissociation kinetics of the longer 2′-OMe-RNA probes might be slower, since the incremental affinity loss induced upon end fraying (caused by the dissociation of a base pair at the terminus) is smaller than with the shorter PNA probes, which provide higher stability per base pair. It may also be that 2′-OMe-RNA oligonucleotides have generally different hybridization kinetics than PNA. In fact, comparatively low koff values have been reported for other 2′-ribose modified oligonucleotides.[50] Regardless of the causes, the comparison between PNA and RNA probes points to the special properties of PNA that indicate a particular suitability for template-controlled reactions. In addition, PNA generally has a low tolerance for base mismatches. Though not subject of this study, we refer to previous work of our group in which we have demonstrated the high sequence fidelity provided by templated reaction of short PNA probes.[51-58]
4 CONCLUSIONSIn summary, we have developed a system for an RNA-induced reductive activation of azide-modified Smac mimetic peptides. The comparative study revealed that the photoredox catalyst Ru(bpy)(mcbpy)2 provided higher rates and higher reaction yields than the phosphine based reducing agent dmTCEP. Using azidopeptide conjugates and RNA template in equimolar ratios at 37 °C, up to 90% yield of activated mono- or bivalent Smac mimetic could be achieved in 15 to 20 minutes. In the absence of RNA, only 2% to 3% product was formed. The reaction was fastest when a minimal gap of 1 unpaired nucleotide was present between the probe annealing sites on the template, resulting in a rate enhancement over the background reaction of 870-fold and 1100-fold at 25 °C and 37 °C, respectively. A comparison between PNA and Tm-adjusted 2′-OMe-RNA probes underscored the unique properties that reactive PNA conjugates provide in template-controlled chemistry. The PNA probes were chemically more stable against photoinduced side reactions and provided higher reaction rates/yields when the reactions were performed under turnover conditions. On 10 nM (0.01 eq.) target RNA, the photoreduction reaction provided 130 nM Smac mimetic conjugate. Such an RNA concentration may be realistically achieved by target overexpressing cells, making the reaction suitable for future evaluation in a biological system, for example, the highly SMC-sensitive MDA-MB-231 or EVSA-T human breast cancer cell lines. Since visible light is used for excitation of the catalyst, the irradiation procedure should be well tolerated. In fact, Winssinger and coworkers, have recently demonstrated that a similar, ruthenium-based reaction system could be employed for a fluorogenic detection of miRNA in living cells and in vivo.[27, 38] Although PNA is more difficult to deliver into cells than negatively charged 2′-OMe-RNA, this limitation may be overcome with physical transfection techniques, like electroporation. An alternative could be positively charged guanidinium-PNA, which other groups have successfully used for probe delivery.[
留言 (0)