
記住我
Multiple sclerosis (MS) is a chronic autoimmune disease that leads to inflammation, demyelination, and neurodegeneration in the central nervous system (CNS) (1). The primary pathology underlying this disease is axonal demyelination. Lesion location and burden contribute to the variable clinical presentation and disease severity seen in this population. The disease is more common in women than in men, with a ratio of about 3:1, and in individuals of Northern European descent (2). In fact, MS is one of the most common autoimmune neurologic conditions worldwide with about 2.9 million individuals diagnosed (3). While optic neuritis (ON) can commonly be seen in MS, co-morbid mitochondrial eye disease has also been described in this population.
Leber hereditary optic neuropathy (LHON) is a mitochondrial disorder characterized by bilateral central vision loss due to degeneration of retinal ganglion cells and the optic nerves. It is most commonly seen in young males (4–6). Several mitochondrial DNA variants which disrupt complex I of the respiratory chain have been associated with the disease (6–9). Interestingly, these variants have incomplete penetrance with only about 50% of males and 10% of females with these mutations ultimately developing the condition. This suggests additional genetic, epigenetic, and environmental factors may be involved (6, 10). In addition to the complex genetics at play, mutations may also be synergistic in some cases, producing specific phenotypes (11). As there are no definitive or curative treatments for LHON, the condition is considered neurodegenerative and progressive in most cases, although emerging interventions such as gene therapy are on the horizon (12, 13).
Previous reports have described cases of LHON and MS co-occurrence. This overlap is predominantly seen in female patients (14). It has been proposed that harboring LHON variants is a risk factor for developing MS (15). It is possible that mitochondrial DNA variants in persons with MS (PwMS) could potentially contribute to unique clinical subgroups, emphasizing the need for further investigation into the overlapping pathophysiology of LHON and MS (16). This narrative review will explore the complex interactions between mitochondrial disease and neuroinflammation to enhance our understanding of the shared mechanisms underlying these disorders.
2 Clinical and radiographic overlap in LHON and MSThe overlap between LHON and MS has gained attention in recent days, such that LHON-MS has been given a distinct name, Harding’s Disease (17). This association was initially reported by Lees et al. in 1964, who observed that LHON and MS could co-occur in the same individual (18). Harding et al. further expanded on this observation, documenting 11 cases in which LHON and MS symptoms coexisted, supporting the idea that LHON mutations might predispose individuals to neuroinflammation and associated conditions such as MS (17).
Recent studies support this association. An extensive review involving 55 LHON families and 40 patients with confirmed MS highlighted that primary LHON mutations are associated with an increased risk of developing MS (15). Notably, all three primary LHON mutations identified in European and North American populations (m.11778A > G, m.3460A > G, and m.14484 T > C) have been associated with symptoms similar to those of MS, including vision problems, motor function issues, and cognitive difficulties, suggesting a genetic predisposition (15). This association is particularly evident in females despite LHON predominantly affecting men, indicating that MS may be triggered in women with LHON when certain environmental factors are present (14, 18).
LHON-MS shares certain features with MS including age of onset, a female predilection, and a predominance of the relapsing–remitting MS phenotype (71.1%). However, it differs significantly in that 96% of LHON-MS patients experience visual involvement with only 10% reporting ocular pain and 72.1% lacking visual recovery, resulting in 50% of patients registering as legally blind. In contrast, only 50% of patients with isolated MS have visual involvement with 85 to 95% recovering to better than 6/9 visual acuity (19–21). These features may be important when differentiating these disease processes as painless ON and lack of visual improvement with treatment would be atypical for isolated neuroinflammatory disorders (Table 1). This suggests that LHON-MS has a distinct clinical phenotype that may reflect a unique mechanistic interaction between LHON and MS.

Table 1. Clinical differences between LHON, ON, LHON-MS, NMOSD, and typical MS (6, 17, 19).
Neuroimaging studies show that the imaging characteristics of LHON-MS closely resemble those of MS (Figure 1) (14, 17). Per McDonald’s 2017 criteria, neuroimaging abnormalities in MS typically occur in multiple areas of the CNS, showing patterns of indistinguishable white matter lesions and brain atrophy, which have also been reported in individuals with LHON-MS (14, 22). As compared to LHON alone, neuroimaging in patients with LHON-MS demonstrates more extensive white matter abnormalities and optic nerve damage (23).
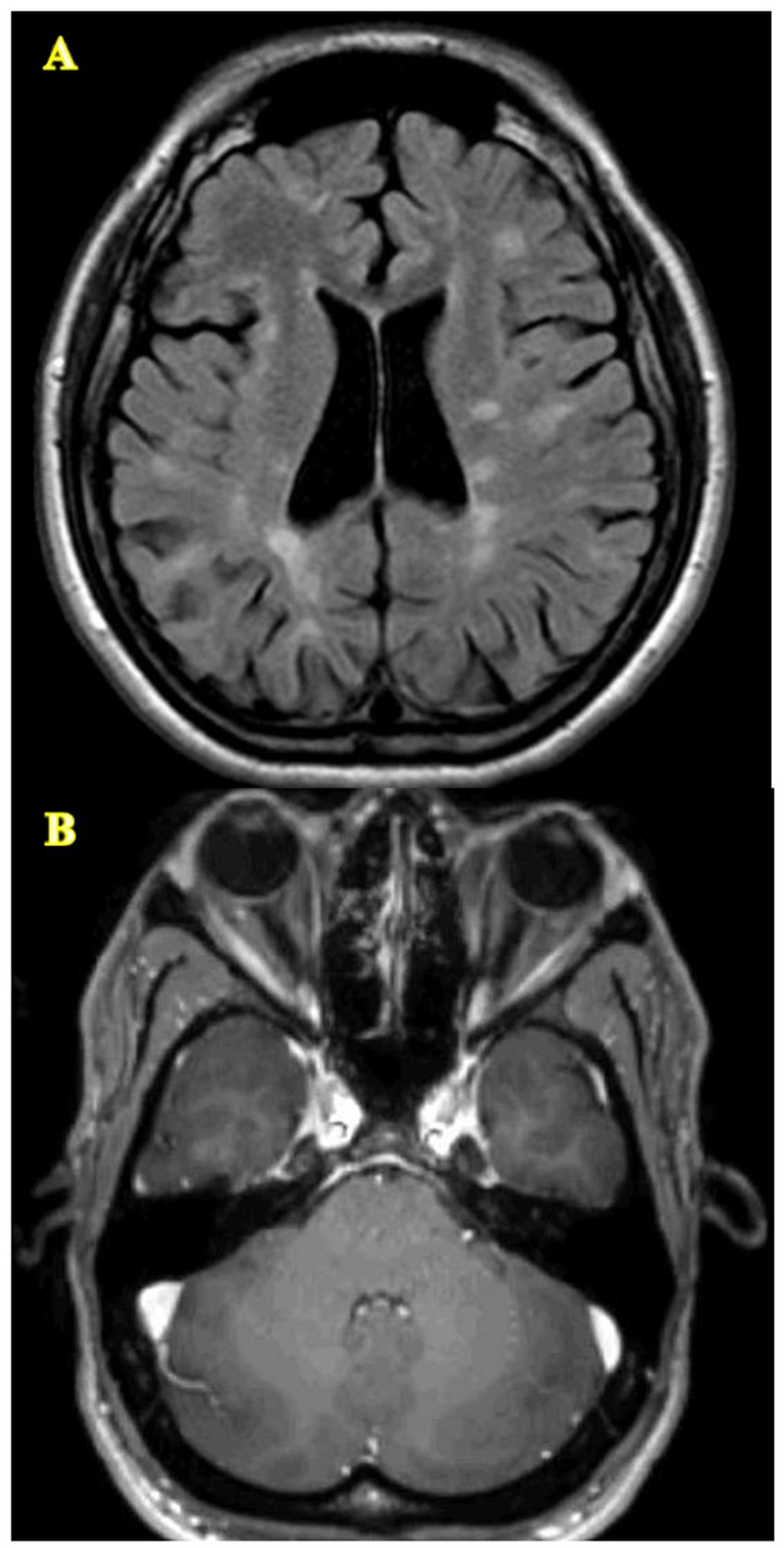
Figure 1. Lesions of the CNS characteristic of LHON-MS. (A) Diffuse T2/FLAIR signal abnormalities throughout the white matter meeting McDonald’s 2017 criteria. (B) Atrophy of the optic nerves.
3 LHON and mitochondrial dysfunctionLHON is a mitochondrial disorder that leads to severe visual impairment or blindness due to the degeneration of retinal ganglion cells (RGCs). The disease is mainly caused by point mutations in mitochondrial DNA (mtDNA) affecting the complex I subunits of the electron transport chain, which leads to less effective cellular energy production and higher susceptibility to cell death (24). The typical clinical presentation includes subacute bilateral visual loss, central scotoma, dyschromatopsia, and eventual optic disk atrophy, with early stages marked by pseudoedema and microangiopathy (6, 25).
The role of mitochondrial dysfunction in the pathogenesis of LHON is well established. Ghelli et al. demonstrated that osteosarcoma-derived cytoplasmic hybrids (cybrids) with common LHON mutations (11,778/ND4, 3,460/ND1, and 14,484/ND6) undergo apoptotic cell death when under metabolic stress induced by galactose, which leads cells to rely on mitochondrial respiration for ATP production (26). This study showed that LHON cybrids exhibit signs of apoptosis such as chromatin condensation, nuclear DNA laddering, and increased cytochrome c release into the cytosol, with mutations 3,460/ND1 and 14,484/ND6 leading to greater apoptotic susceptibility compared to 11,778/ND4 (Figure 2) (24).
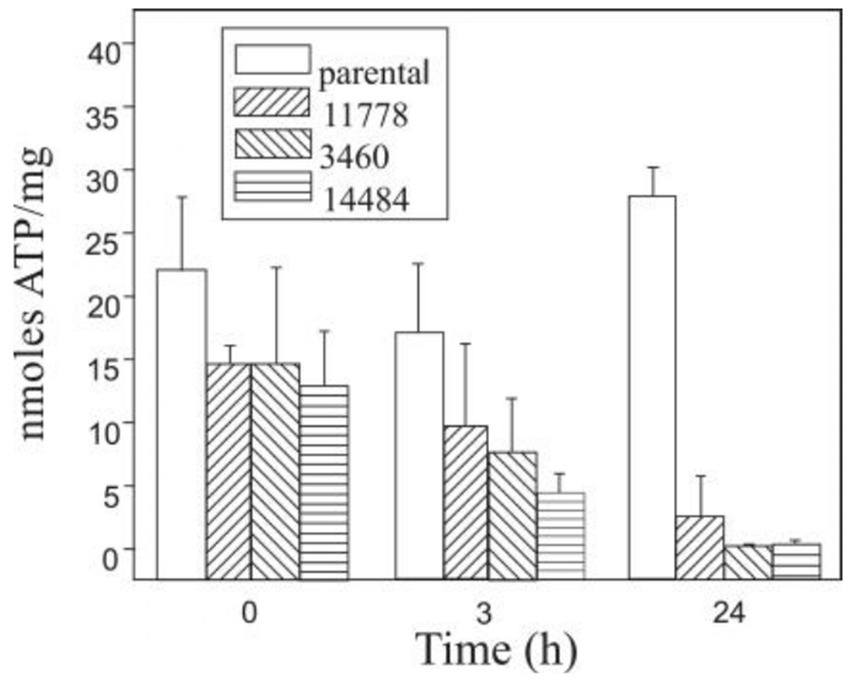
Figure 2. ATP content in 142B.TK- cybrid cells, harboring the three most common primary LHON mutations, and in the parental cell line were determined in triplicate after various incubation periods in galactose-medium (27).
In addition to mitochondrial dysfunction, the regulation of superoxide production in RGCs compared to brain and neuroblastoma cells has been explored. Research by Levin found that RGCs produce superoxide at lower rates than brain mitochondria with tighter regulation, potentially preventing aberrant apoptosis signaling (25). Disruption of this balance by LHON-related mtDNA mutations could lead to increased superoxide levels, contributing to RGC death and optic neuropathy (25, 27).
The complexity of diagnosing LHON is in part due to its similarity to other conditions such as neuromyelitis optica spectrum disorders (NMOSD), isolated ON, and MS. Accurate diagnosis relies on high clinical suspicion and comprehensive diagnostic resources (Table 1), with definitive diagnosis achieved through identifying specific mtDNA mutations (Table 2) (28). Recent findings have also identified new autosomal recessive mutations in patients with LHON-like symptoms, broadening the spectrum of the disease (29).
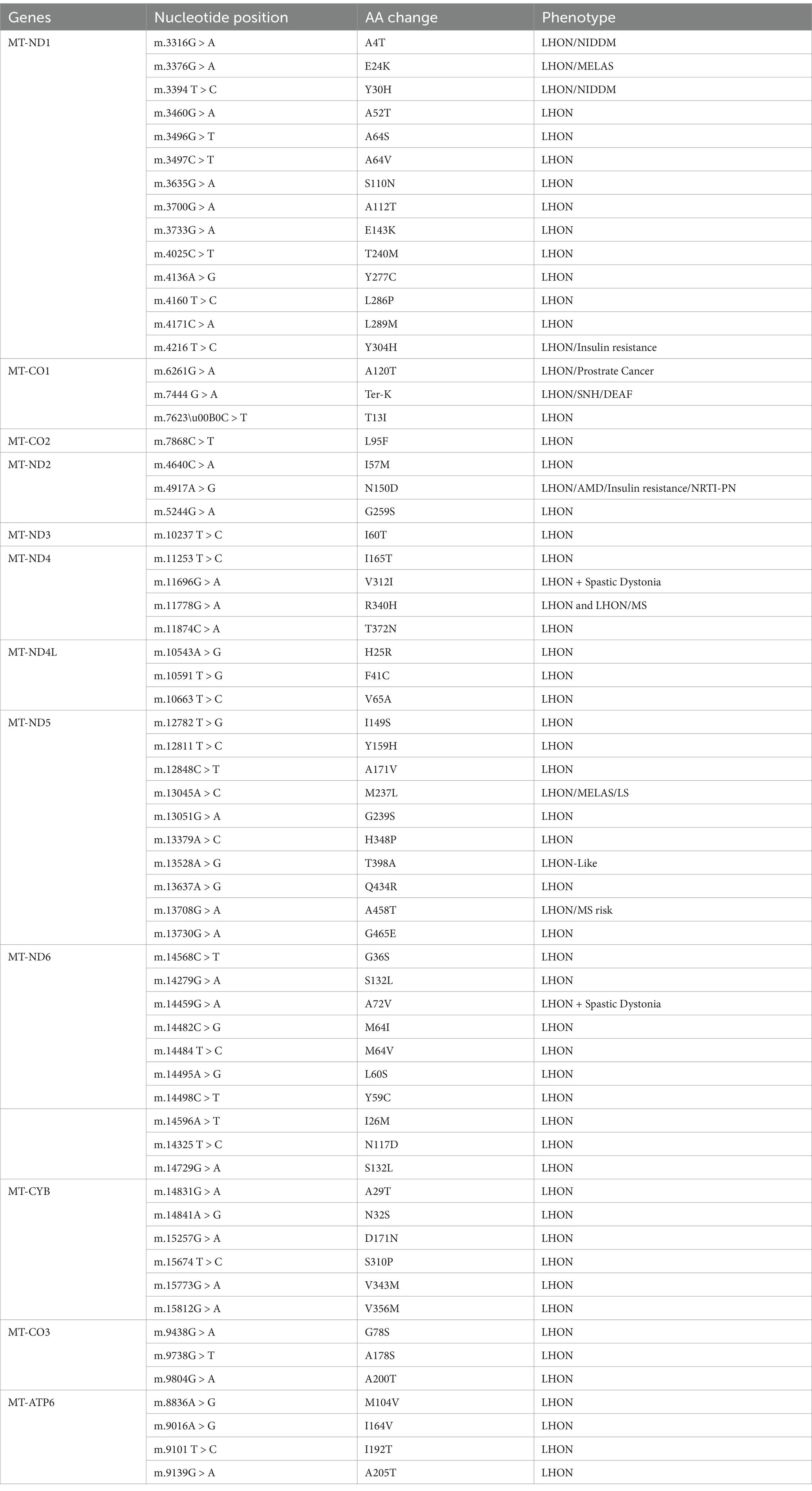
Table 2. Mitochondrial DNA variants identified in individuals with LHON (13).
Inflammation is initiated through the activation of pattern recognition receptors (PRRs) present in both immune and non-immune cells, which respond to microbial elements and endogenous signals known as damage-associated molecular patterns (DAMPs) (30). Under normal physiological conditions, DAMPs, including ATP and specific proteins, remain sequestered within cellular compartments and are unable to activate PRRs. However, cellular stress or death can alter membrane permeability, thereby facilitating the release of DAMPs and the subsequent initiation of inflammatory responses (30). Mitochondria play a pivotal role in this process for several reasons: they possess evolutionary similarities to bacteria indicating a potential interaction with PRRs; their dual membrane structure allows for the regulation of mitochondrial DAMP release; and they are crucial in mediating various forms of regulated cell death that promote DAMP redistribution and PRR activation (31–33). Thus, mitochondria serve as integral components in the regulation of inflammation, linking cellular stress responses to immune activation and contributing to the maintenance of homeostasis (34, 35). As such, it remains possible that in individuals with LHON-MS, mitochondrial dysfunction may be triggering a secondary immune phenomenon.
4 Possible mitochondrial dysfunction in MSMitochondria play a key role in the generation of cellular energy and various metabolic functions in neurons and oligodendrocytes (36). Mitochondria in oligodendrocytes play a vital role in providing the necessary energy for myelin synthesis, which is critical for proper neuronal function (36). They also contribute to controlling lactate availability, which helps with axonal function. Furthermore, mitochondria regulate calcium levels and have the ability to induce apoptosis when there is an excess of calcium, especially during periods of ischemia (36). A recent study used fluorescent markers in mitochondria to gain understanding of how they behave dynamically in the myelin sheath (36). These markers revealed that mitochondria are spread out in the myelin sheath, with the greatest concentration located in the cytoplasmic ridges beside the axon (36, 37). This distribution indicates that mitochondria play a specific role in maintaining the integrity of myelin and the health of axons (38). Electron microscopy has shown that mitochondria in oligodendrocytes have a smaller cristae surface area than those in neurons, suggesting a decreased capacity for ATP production (36). The results emphasize the significance of mitochondrial function in sustaining the energy demands of oligodendrocytes and supporting cognitive functions while also providing a potential neuroprotective therapeutic avenue for future study (39, 40).
Mitochondrial dysfunction is increasingly recognized as a critical factor in the pathophysiology of MS (41, 42). The depletion of myelin results in heightened energy requirements for neurons, due to the extensive participation of the axonal membrane in depolarization, resulting in elevated ATP usage (43). The heightened need may surpass the mitochondria’s capability to generate ATP, leading to compromised neuronal function. Mitochondria play a key role in producing ATP through oxidative phosphorylation (44). In MS, their impairment results in lower ATP production and increased oxidative stress (45). Research has shown that there is a higher amount of mitochondria and enzyme activity, specifically complex IV, in MS lesions than in normal-appearing white matter (46). Additionally, there is increased expression of mitochondrial stress proteins such as mtHSP70 in these regions. The neuroinflammatory environment characteristic of MS is also associated with increased production of reactive oxygen species (ROS), which are partly generated by dysfunctional mitochondria (47). This oxidative stress exacerbates inflammation and contributes to axonal damage and degeneration (48). These observations suggest that mitochondrial stress and oxidative damage are significant contributors to tissue damage and disease progression in MS (46). These pathological processes seem to yield a destructive potentiation cascade wherein further tissue damage also further impairs mitochondrial activity which has been directly associated with clinical disease progression (49). Moreover, mitochondrial inhibitors like rotenone have been shown to impair oligodendrocyte differentiation, further linking mitochondrial dysfunction to demyelination in MS (50).
These studies provide evidence that individuals with genetic disorders predisposing them to mitochondrial dysfunction may face an increased risk of developing MS or experiencing more severe disease manifestations. As inflammation has a detrimental impact on mitochondrial function, those with preexisting mitochondrial impairments could be more susceptible to the cellular damage seen in inflammatory processes such as MS and therefore have more severe disease presentation and rate of progression. While MS is not inherently a mitochondrial disorder, its neuroinflammatory processes can lead to mitochondrial dysfunction, a situation that may differ in LHON-MS, in which mitochondrial dysfunction is likely occurring in a bidirectional manner. These findings point to the critical role of mitochondrial health in MS severity and progression and highlight the potential of therapeutic targets aimed at mitigating mitochondrial dysfunction and oxidative damage (51, 52). Further studies are required to better characterize the impact of underlying mitochondrial dysfunction on clinical presentation and outcomes in patients with MS.
5 Therapeutic advancement in LHONRecent treatment advancements for LHON have been targeted at treating mitochondrial dysfunction, which is fundamental to the disease’s pathology due to the impaired mitochondrial function in RGCs (53–55). Antioxidants such as Idebenone, a synthetic analog of coenzyme Q10, aim to enhance mitochondrial bioenergetics and mitigate oxidative stress (54). While initial studies suggested potential benefits in visual acuity (56), larger clinical trials such as the RHODOS study have produced mixed results (57). These findings highlight the complexity of mitochondrial involvement in LHON and elucidate the need for a deeper understanding of how mitochondrial dysfunction contributes to visual impairment.
Gene therapy has emerged as a promising approach, utilizing strategies such as allotopic expression to relocate mitochondrial genes to the nucleus which facilitates proper protein synthesis within mitochondria (58, 59). Early clinical trials targeting the ND4 mutation have shown some improvements in vision, suggesting that restoring mitochondrial function can yield positive outcomes, although with variable responses among patients (60). Additionally, emerging nutritional interventions such as the ketogenic diet aim to strengthen mitochondrial bioenergetics. Mitochondrial replacement therapy (MRT) also holds promise for preventing the transmission of LHON mutations by combining healthy mitochondrial DNA with nuclear DNA from affected individuals, although ethical considerations and technical challenges remain significant (61, 62). Given the potential curative role of these treatments and the ability to affect multiple areas of the body simultaneously, significant research efforts are likely in this area in the next few years (63, 64). Recent studies have also hypothesized that mitochondrial transplantation, which involves transferring healthy mitochondria into damaged cells, may be an effective treatment for MS (64).
6 Treatment overlap in LHON-MSDisease-modifying agents for MS have never been tested for effectiveness in long-term disability reduction in LHON, as the established pathology of MS is likely distinct from LHON. Idebenone and Mitoxantrone have shown some benefit in LHON. Idebenone is considered safe and has led to visual improvements in some LHON cases (19, 57). Mitoxantrone has also shown some benefit but carries significant risks, making it less favorable compared to safer alternatives (65). On the other hand, LHON-MS may overlap significantly from both a clinical and radiographic standpoint with MS alone. Given the high likelihood of severe visual disability in LHON-MS patients, early intervention with disease-modifying therapies is recommended. Additionally, recent studies suggest that 4-aminopyridine may improve visual evoked potentials in certain MS patients, making it a potential candidate for further research in the LHON-MS population (66).
7 Future directions and research gapsDespite emerging evidence linking mitochondrial dysfunction to both MS and LHON, significant gaps remain in understanding the specific roles of mitochondria in these conditions. Key areas where research is lacking include the precise molecular mechanisms by which mitochondrial dysfunction contributes to neuronal damage in MS and the interplay of genetic and environmental factors that may influence these processes. Furthermore, the extent to which mitochondrial mutations in LHON could predispose individuals to MS-like symptoms needs more investigation, particularly in diverse populations beyond those previously studied. Future studies should prioritize longitudinal investigations into mitochondrial dysfunction, assessing how these abnormalities evolve over time in MS and LHON patients.
8 ConclusionThe overlap of MS and LHON shines light on a complex relationship in which mitochondrial dysfunction plays a central role. The co-occurrence of these two conditions suggests that LHON mutations may predispose individuals to MS and possibly contribute to more severe clinical manifestations in patients. Both diseases exhibit neuroinflammation, visual impairment, and white matter lesions, highlighting the need for further research into how mitochondrial dysfunction influences disease progression. Understanding the mechanisms by which LHON mutations impact MS development remains an important area for future research, as it could lead to mitochondrial targeted interventions. As our understanding of mitochondrial involvement in these diseases grows, we will become closer to developing more effective, tailored therapies for patients suffering from both MS and LHON.
Author contributionsGR: Conceptualization, Formal analysis, Methodology, Project administration, Writing – original draft. MS: Conceptualization, Formal analysis, Writing – original draft, Writing – review & editing. ML: Formal analysis, Writing – original draft, Writing – review & editing. LK: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. MY: Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. SJ: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. JS: Conceptualization, Formal analysis, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References2. Koch-Henriksen, N, and Sørensen, PS. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. (2010) 9:520–32. doi: 10.1016/S1474-4422(10)70064-8
Crossref Full Text | Google Scholar
3. Graf, J, Akmatov, MK, Meuth, SG, Tremlett, H, and Holstiege, J. Updated multiple sclerosis incidence, 2015-2022. JAMA Neurol. (2024) 81:1100–2. doi: 10.1001/jamaneurol.2024.2876
PubMed Abstract | Crossref Full Text | Google Scholar
5. Takano, F, Ueda, K, Godefrooij, DA, Yamagami, A, Ishikawa, H, Chuman, H, et al. Incidence of Leber hereditary optic neuropathy in 2019 in Japan: a second nationwide questionnaire survey. Orphanet J Rare Dis. (2022) 17:319. doi: 10.1186/s13023-022-02478-4
PubMed Abstract | Crossref Full Text | Google Scholar
6. Yu-Wai-Man, P, Griffiths, PG, and Chinnery, PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res. (2011) 30:81–114. doi: 10.1016/j.preteyeres.2010.11.002
PubMed Abstract | Crossref Full Text | Google Scholar
7. Yuan, J, Zhao, J, Ye, C, Pang, L, Zhang, X, Luk, A, et al. Leber's hereditary optic neuropathy with mitochondrial DNA mutation G11778A: a systematic literature review and Meta-analysis. Biomed Res Int. (2023) 2023:1107866. doi: 10.1155/2023/1107866
PubMed Abstract | Crossref Full Text | Google Scholar
8. Guo, DY, Wang, XW, Hong, N, and Gu, YS. A Meta-analysis of the association between different genotypes (G11778A, T14484C and G3460A) of Leber hereditary optic neuropathy and visual prognosis. Int J Ophthalmol. (2016) 9:1493–8. doi: 10.18240/ijo.2016.10.21
PubMed Abstract | Crossref Full Text | Google Scholar
9. Shafa Shariat Panahi, M, Houshmand, M, and Tabassi, AR. Mitochondrial D-loop variation in leber hereditary neuropathy patients harboring primary G11778A, G3460A, T14484C mutations: J and W haplogroups as high-risk factors. Arch Med Res. (2006) 37:1028–33. doi: 10.1016/j.arcmed.2006.04.009
PubMed Abstract | Crossref Full Text | Google Scholar
10. Emperador, S, Habbane, M, López-Gallardo, E, del Rio, A, Llobet, L, Mateo, J, et al. Identification and characterization of a new pathologic mutation in a large Leber hereditary optic neuropathy pedigree. Orphanet J Rare Dis. (2024) 19:148. doi: 10.1186/s13023-024-03165-2
PubMed Abstract | Crossref Full Text | Google Scholar
11. Achilli, A, Iommarini, L, Olivieri, A, Pala, M, Hooshiar Kashani, B, Reynier, P, et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber's hereditary optic neuropathy. PLoS One. (2012) 7:e42242. doi: 10.1371/journal.pone.0042242
PubMed Abstract | Crossref Full Text | Google Scholar
12. Shamsnajafabadi, H, MacLaren, RE, and Cehajic-Kapetanovic, J. Current and future landscape in genetic therapies for Leber hereditary optic neuropathy. Cells. (2023) 12:2013. doi: 10.3390/cells12152013
PubMed Abstract | Crossref Full Text | Google Scholar
13. Koilkonda, RD, Yu, H, Chou, TH, Feuer, WJ, Ruggeri, M, Porciatti, V, et al. Safety and effects of the vector for the Leber hereditary optic neuropathy gene therapy clinical trial. JAMA Ophthalmol. (2014) 132:409–20. doi: 10.1001/jamaophthalmol.2013.7630
PubMed Abstract | Crossref Full Text | Google Scholar
14. Matthews, L, Enzinger, C, Fazekas, F, Rovira, A, Ciccarelli, O, Dotti, MT, et al. MRI in Leber's hereditary optic neuropathy: the relationship to multiple sclerosis. J Neurol Neurosurg Psychiatry. (2015) 86:537–42. doi: 10.1136/jnnp-2014-308186
PubMed Abstract | Crossref Full Text | Google Scholar
15. Vanopdenbosch, L, Dubois, B, D'Hooghe, MB, Meire, F, and Carton, H. Mitochondrial mutations of Leber's hereditary optic neuropathy: a risk factor for multiple sclerosis. J Neurol. (2000) 247:535–43. doi: 10.1007/s004150070153
PubMed Abstract | Crossref Full Text | Google Scholar
16. Alorainy, J, Alorfi, Y, Karanjia, R, and Badeeb, N. A comprehensive review of Leber hereditary optic neuropathy and its association with multiple sclerosis-like phenotypes known as Harding's disease. Eye Brain. (2024) 16:17–24. doi: 10.2147/EB.S470184
PubMed Abstract | Crossref Full Text | Google Scholar
17. Harding, A, Sweeney, M, Miller, D, Mumford, C, Kellar-Wood, H, Menard, D, et al. Occurrence of a multiple sclerosis-like illness in women who have a Leber's hereditary optic neuropathy mitochondrial DNA mutation. Brain. (1992) 115:979–89. doi: 10.1093/brain/115.4.979
PubMed Abstract | Crossref Full Text | Google Scholar
18. Lees, F, Macdonald, AM, and Turner, JW. Leber's disease with symptoms resembling disseminated sclerosis. J Neurol Neurosurg Psychiatry. (1964) 27:415–21. doi: 10.1136/jnnp.27.5.415
PubMed Abstract | Crossref Full Text | Google Scholar
19. Pfeffer, G, Burke, A, Yu-Wai-Man, P, Compston, DA, and Chinnery, PF. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology. (2013) 81:2073–81. doi: 10.1212/01.wnl.0000437308.22603.43
PubMed Abstract | Crossref Full Text | Google Scholar
20. McDonald, WI, and Barnes, D. The ocular manifestations of multiple sclerosis. 1. Abnormalities of the afferent visual system. J Neurol Neurosurg Psychiatry. (1992) 55:747–52. doi: 10.1136/jnnp.55.9.747
PubMed Abstract | Crossref Full Text | Google Scholar
21. The clinical profile of optic neuritis. Experience of the optic neuritis treatment trial. Optic Neuritis Study Group. Arch Ophthalmol. (1991) 109:1673–8. doi: 10.1001/archopht.1991.01080120057025
Crossref Full Text | Google Scholar
22. Thompson, AJ, Banwell, BL, Barkhof, F, Carroll, WM, Coetzee, T, Comi, G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. (2018) 17:162–73. doi: 10.1016/S1474-4422(17)30470-2
PubMed Abstract | Crossref Full Text | Google Scholar
23. Inglese, M, Rovaris, M, Bianchi, S, Comi, G, and Filippi, M. Magnetization transfer and diffusion tensor MR imaging of the optic radiations and calcarine cortex from patients with Leber's hereditary optic neuropathy. J Neurol Sci. (2001) 188:33–6. doi: 10.1016/S0022-510X(01)00542-1
Crossref Full Text | Google Scholar
25. Levin, LA. Mechanisms of retinal ganglion specific-cell death in Leber hereditary optic neuropathy. Trans Am Ophthalmol Soc. (2007) 105:379–91.
PubMed Abstract | Google Scholar
26. Ghelli, A, Zanna, C, Porcelli, AM, Schapira, AHV, Martinuzzi, A, Carelli, V, et al. Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J Biol Chem. (2003) 278:4145–50. doi: 10.1074/jbc.M210285200
PubMed Abstract | Crossref Full Text | Google Scholar
29. Lenaers, G, Beaulieu, C, Charif, M, Gerber, S, Kaplan, J, and Rozet, JM. Autosomal recessive Leber hereditary optic neuropathy, a new neuro-ophthalmo-genetic paradigm. Brain. (2023) 146:3156–61. doi: 10.1093/brain/awad131
PubMed Abstract | Crossref Full Text | Google Scholar
32. Dela Cruz, CS, and Kang, MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. (2018) 41:37–44. doi: 10.1016/j.mito.2017.12.001
PubMed Abstract | Crossref Full Text | Google Scholar
33. Zhang, Q, Raoof, M, Chen, Y, Sumi, Y, Sursal, T, Junger, W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
PubMed Abstract | Crossref Full Text | Google Scholar
35. de, A, Solodka, K, Zanini, G, Selleri, V, Mattioli, A, Nasi, M, et al. Molecular mechanisms of mtDNA-mediated inflammation. Cells. (2021) 10:11. doi: 10.3390/cells10112898
PubMed Abstract | Crossref Full Text | Google Scholar
36. Rinholm, JE, Vervaeke, K, Tadross, MR, Tkachuk, AN, Kopek, BG, Brown, TA, et al. Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia. (2016) 64:810–25. doi: 10.1002/glia.22965
PubMed Abstract | Crossref Full Text | Google Scholar
37. Edgar, JM, McCulloch, MC, Thomson, CE, and Griffiths, IR. Distribution of mitochondria along small-diameter myelinated central nervous system axons. J Neurosci Res. (2008) 86:2250–7. doi: 10.1002/jnr.21672
PubMed Abstract | Crossref Full Text | Google Scholar
38. Ohno, N, Kidd, GJ, Mahad, D, Kiryu-Seo, S, Avishai, A, Komuro, H, et al. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J Neurosci. (2011) 31:7249–58. doi: 10.1523/JNEUROSCI.0095-11.2011
PubMed Abstract | Crossref Full Text | Google Scholar
39. Licht-Mayer, S, Campbell, GR, Canizares, M, Mehta, AR, Gane, AB, McGill, K, et al. Enhanced axonal response of mitochondria to demyelination offers neuroprotection: implications for multiple sclerosis. Acta Neuropathol. (2020) 140:143–67. doi: 10.1007/s00401-020-02179-x
PubMed Abstract | Crossref Full Text | Google Scholar
40. Rosenkranz, SC, Shaposhnykov, AA, Träger, S, Engler, JB, Witte, ME, Roth, V, et al. Enhancing mitochondrial activity in neurons protects against neurodegeneration in a mouse model of multiple sclerosis. eLife. (2021) 10:10. doi: 10.7554/eLife.61798
PubMed Abstract | Crossref Full Text | Google Scholar
41. Ghafourifar, P, Mousavizadeh, K, Parihar, MS, Nazarewicz, RR, Parihar, A, and Zenebe, WJ. Mitochondria in multiple sclerosis. Front Biosci. (2008) 13:3116–26. doi: 10.2741/2913
PubMed Abstract | Crossref Full Text | Google Scholar
42. Dhib-Jalbut, S, Arnold, DL, Cleveland, DW, Fisher, M, Friedlander, RM, Mouradian, MM, et al. Neurodegeneration and neuroprotection in multiple sclerosis and other neurodegenerative diseases. J Neuroimmunol. (2006) 176:198–215. doi: 10.1016/j.jneuroim.2006.03.027
PubMed Abstract | Crossref Full Text | Google Scholar
44. Kiryu-Seo, S, Ohno, N, Kidd, GJ, Komuro, H, and Trapp, BD. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci. (2010) 30:6658–66. doi: 10.1523/JNEUROSCI.5265-09.2010
PubMed Abstract | Crossref Full Text | Google Scholar
45. Mahad, DJ, Ziabreva, I, Campbell, G, Lax, N, White, K, Hanson, PS, et al. Mitochondrial changes within axons in multiple sclerosis. Brain. (2009) 132:1161–74. doi: 10.1093/brain/awp046
PubMed Abstract | Crossref Full Text | Google Scholar
46. Witte, ME, Bø, L, Rodenburg, RJ, Belien, JA, Musters, R, Hazes, T, et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol. (2009) 219:193–204. doi: 10.1002/path.2582
PubMed Abstract | Crossref Full Text | Google Scholar
47. Chinopoulos, C, Tretter, L, Rozsa, A, and Adam-Vizi, V. Exacerbated responses to oxidative stress by an Na(+) load in isolated nerve terminals: the role of ATP depletion and rise of [ca(2+)](i). J Neurosci. (2000) 20:2094–103. doi: 10.1523/JNEUROSCI.20-06-02094.2000
PubMed Abstract | Crossref Full Text | Google Scholar
49. Armon-Omer, A, Neuman, H, Sharabi-Nov, A, and Shahien, R. Mitochondrial activity is impaired in lymphocytes of MS patients in correlation with disease severity. Mult Scler Relat Disord. (2020) 41:102025. doi: 10.1016/j.msard.2020.102025
留言 (0)