
記住我
Hereditary spherocytosis (HS) is a prevalent genetic disorder characterized primarily by hemolytic anemia, jaundice, splenomegaly, spherocytosis, and a family history of hemolytic anemia. Because of abnormalities in the coding of red blood cell membrane skeleton protein genes, the corresponding membrane skeleton protein synthesis is reduced or defective, leading to partial loss of the red blood cell membrane and the red blood cells becoming spherical. They are then sequestered and phagocytosed by the spleen for clearance (1). Pathogenic variation mainly included the ANK1, SPTA1, SPTB, SLC4A1, and EPB42 genes, encoding ankyrin, α-spectrin, β-spectrin, band 3 protein, and 4.2 protein, respectively (2, 3). Diagnosis of HS typically relies on clinical manifestations, family history, and a series of laboratory tests, which may present false-negative and false-positive results. The clinical manifestations of HS vary greatly, spanning from asymptomatic to severe symptoms that may pose life-threatening risks. Some children with mild disease may not be diagnosed until the occurrence of infections, and others are diagnosed serendipitously during treatment for other diseases. Therefore, the diagnosis of HS poses a challenge, especially for asymptomatic or atypical cases that rely solely on clinical presentation, family history, and hematologic laboratory tests.
With the widespread application of genetic diagnostic technologies, genetic testing offers enhanced diagnostic efficiency and rapidly provides a comprehensive and detailed genetic analysis in patients suspected of HS. Furthermore, early genetic testing can aid in the understanding of phenotypes and inheritance patterns, which also helps in risk assessment and genetic counseling. Initial studies suggested that genetic heterogeneity forms the basis of clinical heterogeneity (4). However, studies on HS in recent decades have been controversial, with most studies suggesting that pediatric patients with SPTA1 variants exhibit more severe anemia, while patients with SLC4A1 mutations present milder phenotypes, such as higher hemoglobin levels and lower reticulocyte counts (5, 6). Researchers from the Netherlands argue that variants in ANK1 and SPTB may lead to heightened severity of HS manifestations in contrast to SPTA1, with these patients typically having lower hemoglobin and higher reticulocyte levels. However, no significant difference between ANK1 and SPTB was observed (7). Research in China on this topic remains scarce, especially in children (8, 9). HS prevalence in China is approximately 1.27 per 100,000 in males and 1.49 per 100,000 in females, most of whom are diagnosed during childhood (10). Due to the significant heterogeneity in clinical manifestations in children with HS, it may be difficult to diagnose those with mild symptoms and those who are frequently transfused with routine laboratory examinations. In this study, we retrospectively summarized and analyzed the clinical and genetic characteristics of 64 Chinese children with HS, investigating the impact of different gene variant types on clinical presentation.
Methods Study participantsThe clinical characteristics of 64 pediatric patients with HS admitted to Beijing Children's Hospital, Capital Medical University from January 2018 to December 2023 were collected for retrospective analysis. Patients were included if they had a clinical diagnosis of HS (based on medical history, non-immune hemolytic anemia, splenomegaly, and spherocytes on morphology) (2) and had genetic testing (HS variation was identified) performed. Informed consent was obtained from the patients' parents. The study was approved by the Ethics Committee of Beijing Children's Hospital.
Data collectionWe retrospectively collected data on patient demographics, family history, data of splenomegaly by abdominal ultrasound, jaundice, anemia, or cholecystolithiasis, laboratory testing data [including blood smears reviewed by hematologist, complete blood count, blood biochemical indexes, and the oxidative fermentative (OF) test (11)], treatments, and therapeutic effects.
Genetic testingAll genetic diagnoses in this study were obtained using targeted-next-generation sequencing (NGS) (a genetic panel of the blood system) or whole-exome sequencing (WES) and were validated using Sanger sequencing. REVEL software was used to predict protein structure-function for unreported novel variants, and pathogenicity analysis was conducted according to the Variant Interpretation Guidelines released by the American College of Medical Genetics and Genomics (ACMG), excluding single nucleotide polymorphisms.
Statistical analysisStatistical analysis was conducted using SPSS software (version 23.0). Categorical data are expressed as frequencies and percentages, and normally distributed continuous data are presented as mean ± standard deviation. Paired t-tests were used to compare normally distributed data, while the Kruskal–Wallis test was used for non-normally distributed continuous data. Pairwise comparisons between groups were performed using the Mann–Whitney U test, with p < 0.05 considered statistically significant.
Results General informationThis study comprised 64 participants, consisting of 34 males and 30 females, with a median age of 3.5 years (range: 0.1–14.6 years). The age of onset ranged from immediately after birth to 12 years, with 46 (72%) having disease onset before 1 year of age. The number of patients who had the disease decreased with increasing age [≥3 years and <7 years, 9 (14%); ≥7 years and <12 years, 3 (5%); ≥12 years, 2 (3%);]. Moreover, 21 patients (33%) had a family history of HS, and 4 of them had parents who underwent a splenectomy. Fifty-five (86%) had a history of neonatal jaundice. Overall, the symptoms were mild, and only four patients underwent exchange transfusion therapy. All patients had undergone abdominal ultrasound. Splenomegaly was present in 49 cases (77%), hepatomegaly in 37 (57%), and cholelithiasis in 9 (14%). Among the 64 patients, approximately two-thirds had received red blood cell transfusions, and 14 (22%, 6 cases of ANK1, and 6 cases of SPTB) developed red blood cell transfusion dependence. In this study, all 64 children had a clinical diagnosis of HS and underwent genetic testing, including 57 using targeted NGS and 7 using WES. The detailed clinical features of these patients with the different genotypes are presented in Table 1.
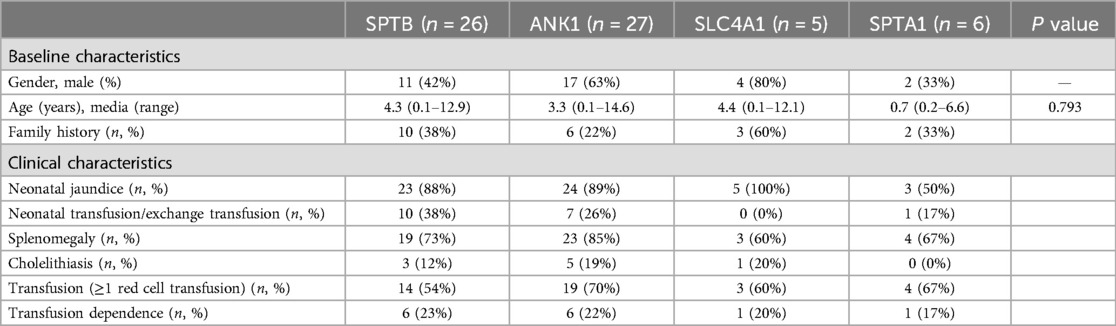
Table 1. Baseline and clinical characteristics of 64 HS children with different gene variants.
Gene variantsVariant data of the 64 patients with HS are shown in Table 2. All patients had heterozygous or compound heterozygous variants. ANK1 and SPTB variants were predominant in all patients, with 27 cases (42%) of ANK1 variants, 26 (41%) of SPTB variants, six (9%) of SPTA1 variants, and five (8%) of SLAC4A1 variants. No EPB42 variants were detected. The sources of variation showed 24 cases (38%) of inherited variants from parents, 35 (55%) were spontaneous, and 5 patients’ fathers or mothers were not sampled. Almost all patients exhibited an autosomal dominant inheritance pattern. In our study, 71 variants were identified, among which 59 variants have not been previously reported. Variation types included 15 cases (21%) of nonsense, 19 (27%) of missense, 28 (39%) of frameshift mutations, 6 (8%) of splicing, and 3 (4%) of large fragment deletions. The ANK1 c.127-3C>G (splicing) variant was found in both patients 17 and 61. Five patients had more than one variant. Secondary SPTA1 variants were the most common (3 cases), but these 5 secondary variants were unlikely to be disease-causing according to ACMG guidelines. Regretfully, for SPTA1, the αLELY allele and αLEPRA allele were not investigated.
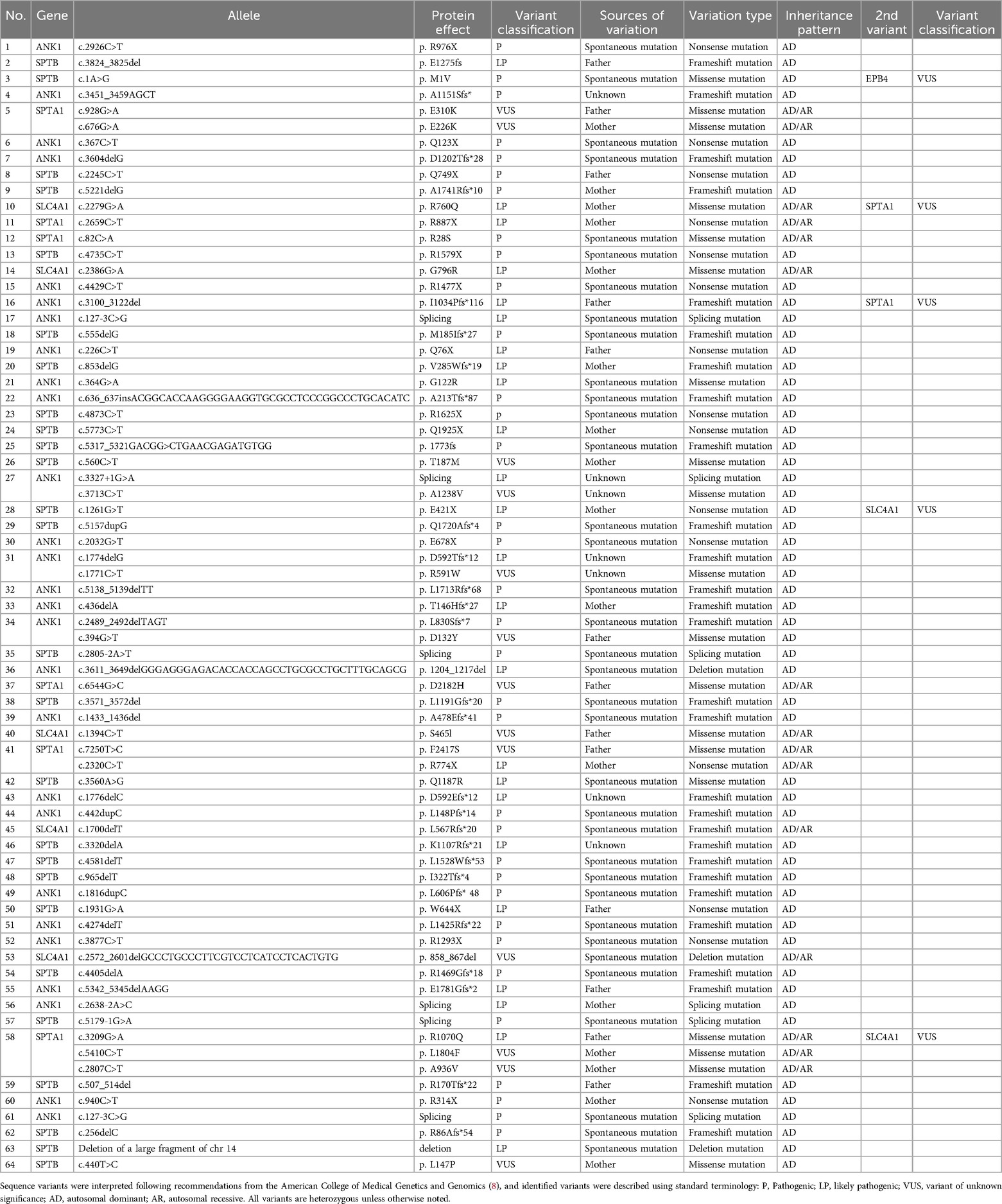
Table 2. Complete list of causative variants in the cohort.
Genotype-phenotype correlationAlmost all patients presented with jaundice, anemia, and splenomegaly, with varying severity. A group analysis of the pathogenicity of different gene variants was performed on 64 patients (Table 3). Spherocytes were observed in the peripheral blood in 21 cases (33%), with >10% spherocytes observed in only one ANK1 case. Anemia was the primary symptom, including mild anemia (Hb 90–120 g/L) in 8 (13%), moderate (Hb 60–90 g/L) in 38 (59%), severe (Hb 30–60 g/L) in 15 (23%), and very severe (Hb <30 g/L) in 1 cases (2%). However, no statistical differences in hemoglobin levels, MCV, MCH, MCHC, or reticulocytes were observed across variant groups. Bilirubin levels were remarkably elevated in patients with HS variants, and those with SPTB-HS had significantly higher bilirubin levels, including total bilirubin (p = 0.033) and indirect bilirubin (p = 0.018) compared to those with SPTA1-HS. A total of 62 HS patients (except for 2 ANK1-HS) completed the traditional OF test. The results showed that the positivity rates in those with SPTB-HS, ANK1-HS, SLC4A1-HS, and SPTA1-HS were 85% (22/26), 88% (22/25), 60% (3/5), and 33% (2/6), respectively. Moreover, ANK1 variants displayed reduced resistance to lysis at varying NaCl concentrations in comparison to those with the SPTA1 variants (p = 0.047). Of the eight patients who underwent splenectomy, five (63%) had ANK1 variants, two (25%) had SPTB variants, and one (12%) had an SLC4A1 variant. None of the patients received splenic embolization or underwent partial splenectomy. The hemoglobin levels of HS patients with SPTB, ANK1 and SLC4A1 variants improved significantly following splenectomy (p < 0.01).
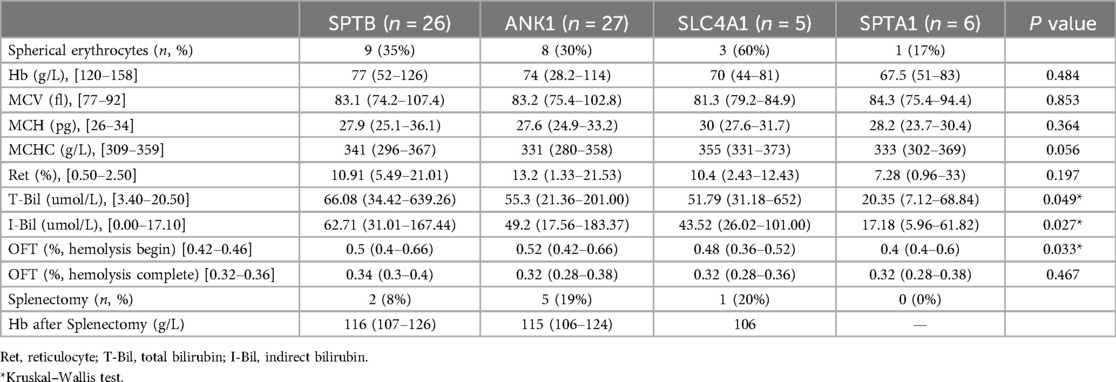
Table 3. Laboratory testing results of 64 HS children with different gene variants.
DiscussionHereditary spherocytosis is based on the pathophysiological effects of defects in genes encoding for one or more of the major RBC cytoskeleton and (trans)membrane proteins: ANK1, SPTB, SPTA1, SLC4A1, and EPB42 (1). HS exhibits great heterogeneity in disease severity among patients, who may be virtually asymptomatic or require frequent transfusions in early childhood. Most patients with HS have mild symptoms, and up to 20%–30% have a purely compensated hemolysis due to a balance between reticulocyte production and red cell destruction (1, 12). Thus, the incidence may be underestimated. In this study, we reported 64 children with HS with an age of disease onset ranging from immediately after birth to 12 years, with >70% of patients having disease onset before 1 year of age. However, the median age at diagnosis was 3.5 years, suggesting that clinical manifestations were not specific and timely to the diagnosis. Conversely, only a third of patients had a family history of HS, also making it difficult to diagnose. Due to the inheritance patterns, some patients had spontaneous mutations, while others showed autosomal recessive inheritance patterns. It is speculated that there may also be incomplete penetrance and variable expressivity among family members (13), so more laboratory tests are needed to diagnose HS.
Currently, the diagnosis of HS mainly relies on the biochemical hemolysis parameters, spherocytes on morphology, and functional testing, such as the OF test, eosin-5-maleimide (EMA) binding test, and membrane protein defects with the SDS-PAGE (14, 15). The drawbacks of the peripheral blood erythrocyte morphology and osmotic fragility test lie in the lack of sensibility and specificity, as other congenital red blood cell defects or hemolytic anemias, may also yield positive results. In our study, spherocytes were observed in the peripheral blood in only a small number of patients, with >10% spherocytes seen in only one ANK1 case. The determination of the membrane skeleton protein involves the extraction of cell membrane proteins using polyacrylamide gel electrophoresis, which is very intricate and has not been widely promoted in clinical practice. The EMA binding test is widely recognized as the most convenient, sensitive, and specific diagnostic method for diagnosing HS. The combination of this test with other red blood cell osmotic tests has been recommended to enhance diagnostic sensitivity (15). However, a shortcoming of the EMA binding test is the lack of normal controls and a universal reference range for HS (16). Existing research on HS indicates that patients with HS who have an ankyrin protein deficiency have low sensitivity in the EMA binding test (17). Similarly, other red blood cell genetic disorders may also show reduced EMA binding levels. Additionally, flow cytometers are not available in all routine diagnostic laboratories, which also restricts their application.
Recently, genetic testing has shown tremendous potential in the diagnosis of HS. Many researchers have reported that patients with HS could benefit from an early diagnosis via genetic testing. In fact, variations in HS-related genes were identified at a high rate, but molecular defects are significantly heterogeneous (6, 18). Autosomal dominant inheritance (AD) and autosomal recessive inheritance (AR) account for 75% and 25% of HS cases, respectively (19), with ANK1, SPTB, and SLC4A1 variants being the most dominant inheritance patterns. Conversely, the SPTA1 and EPB42 defects are often recessive or spontaneous mutations, with ANK1 variants being the most common, followed by SPTB (1, 20). Research on the correlation between genotype and phenotype is still insufficient in HS patients, and it is currently controversial. In this study, there were 27 cases (42%) of ANK1 variants and 26 (41%) of SPTB variants, accounting for over 80% of all variants, which is consistent with previous reports. Since SLC4A1-HS often onsets during adulthood and presents with mild symptoms, the true prevalence of SLC4A1-HS may have been underestimated. Unexpectedly, more than half of the cases were spontaneous mutations and almost all patients exhibited an autosomal dominant inheritance pattern, which may explain the low positive rate of family history. Frameshift, nonsense, and missense mutations are the most common variants. We found 59 variants that have not been reported before. According to the ACMG criteria, 28 novel variants were considered pathogenic, 20 were likely pathogenic, and 11 were unknown. The ANK1 c.127-3C>G and SPTB c.4873C>T were found together in two patients, and the latter has been defined in previous literature as a high-frequency mutation (21).
Herein, we report a significant difference in clinical phenotypes based on the underlying genetic variations. Overall, patients with SPTB-HS and ANK1-HS had the most severe symptoms, while those with SPTA1-HS presented a mild phenotype. Bilirubin levels were remarkably elevated in those with SPTB-HS compared to those with SPTA1-HS. As mentioned above, cholelithiasis was found in 3 patients with SPTB variants, while it was not detected in patients with SPTA1 variants. In addition, ANK1-HS also presented more cholelithiasis and elevated bilirubin levels. This indicates that the level of bilirubin is an important cause of cholelithiasis in these patients, yet it is not the sole determinant. The positive rate of the OFT in SPTB-HS and ANK1-HS was also higher, and ANK1-HS displayed reduced resistance to lysis at varying NaCl concentrations in comparison to SPTA1-HS. Previous studies have shown that certain interactions within the ankyrin complex of the cytoskeleton play a crucial role. Specific variants that interfere with these interactions have been found to lead to significant disruptions in cytoskeleton assembly or function, ultimately resulting in a more pronounced phenotype (22). Additionally, the need for blood transfusions was more common in patients with SPTB and ANK1 variants, especially red blood cell transfusion dependence. Patients with ANK1-HS and SPTB-HS were more likely to undergo splenectomy than other patients. The results of our study are consistent with those of van Vuren, A's research, which reported that variants in ANK1 and SPTB may lead to more severe HS phenotypes compared to variants in SPTA1 (7).
We analyzed variation type and found ANK1 and SPTB non-missense variations increased and might lead to truncated proteins and loss of expression from the affected allele. This disruption of cytoskeleton function was deemed more deleterious than decrease in the quantity of normally formed protein. Besides, the production of α-spectrin was reported to be three to four folds higher than that of β-spectrin in healthy individuals (23, 24). Considering that SPTA1-HS is mostly autosomal recessive owing to dosage compensation effects, most patients with SPTA1-HS only exhibit significant clinical symptoms when homozygous or compound heterozygous mutations occur, resulting in milder phenotype compared to other gene variants. Studies showed patients with low expression of the αLELY allele in SPTA1 generally exhibit milder clinical symptoms even in a homozygous state, making them difficult to identify (25). Another common SPTA1 splicing mutation, where the αLEPRA allele converts to an ineffective variant of SPTA1, results in patients who do not require transfusions and benefit more from splenectomy (26). Conversely, patients with combined mutations of the trans-αLEPRA and αPRAGUE alleles have been reported to have fatal cases (27). This likely explains the phenotypic variability seen in patients with identical pathogenic HS variants is plausibly attributed to the effects of concomitant variants in modifier genes. However, more research is needed to confirm this theory. EPB42 defects are often recessive or spontaneous mutations, and Peters et al. reported EPB42-knockout mice appeared to have the mildest phenotype characterized by a nearly intact membrane skeleton (28). This may be the reason for the low incidence of EPB42-HS.
The distribution of different variant regions in membrane proteins was also considered as a potential factor depending on the severity. Our findings revealed variants distributed across the entire gene. Previous research has indicated that patients with ANK1-HS variants in the spectrin-binding domain exhibit the most severe anemia among affected individuals (22). Moreover, loss-of-function variants in the ZU5 subdomain have also been documented (29, 30). Park et al. suggested that patients with variants in the c.2482-4149 region of the spectrin-binding domain of ANK1 may manifest a more severe phenotype (22). Interestingly, individuals in our cohort did not exhibit a higher severity of anemia despite variant presence in this region. Because no statistical differences across variant groups were observed in hemoglobin levels, MCV, MCH, MCHC, or reticulocytes in our cohort, we conclude that categorization in different genetic subgroups is insufficient to precisely predict HS phenotype. There are several limitations to this study. Firstly, it is a retrospective report. Some patients are transfusion-dependent, which may obscure the laboratory testing results and likely underestimate the severity of the disease. Secondly, it has a small sample size. By increasing the sample size and conducting further research, a more intricate and thorough outcome will be obtained. Thirdly, the incidence of childhood splenectomy may be underestimated in this patient population due to the young age of many participants who may not have had the time to develop complications that would necessitate a splenectomy. Moreover, splenic embolization and partial splenectomy are not available in our center. Stratifying patient age in future studies may help reduce data bias to some extent. In our study, there was a broad phenotypic variability among patients in each genetic subgroup. To further identify the genotype-phenotype correlations in HS, a functional assay of concomitant variants in modifier genes, variant regions, and the pathogenicity of VUS, family members variants, and pedigree analysis are required. New parameters such as mean sphered corpuscular volume (MSCV), Ret/immature reticulocyte fraction (IRF), and mean reticulocyte volume (MRV), were demonstrated to be sensitive parameters for HS detection (31). New insights may be discovered by combining these into diagnostic algorithms and phenotype studies. Although HS is a hereditary disorder cannot be prevented, the progression of molecular diagnostics enables the discovery of new variants and allow for a comprehensive genetic analysis and definitive diagnosis in patients suspected of HS without typical clinical presentation, family history, and hematologic laboratory tests. Through extensive exploration into the relationship between phenotype and genotype in HS, early genetic testing can be utilized in the assessment of disease risk and the selection of appropriate treatments. It can also aid in distinguishing spontaneous mutations and autosomal recessive inheritance cases, thus facilitating genetic counseling for affected families, which may also be beneficial for investigating the specific underlying pathophysiological mechanisms of individuals or families with HS.
ConclusionIn summary, we reported our findings in 64 children with HS with different red cell membrane cytoskeleton protein gene variants and analyzed the clinical phenotypes stratified by variant types, with ANK1 and SPTB variants being associated with the most severe disease and SPTA1 variants with the mildest. To explore the correlation between clinical and mutational features of HS patients, genetic testing is proposed in patients without a family history or who are difficult to diagnose with routine laboratory tests, which may also provide references for clinical treatment and genetic counseling.
Data availability statementThe datasets generated during and/or analysed during the current study are not publicly available. However, datasets are available from the corresponding author on reasonable request.
Ethics statementThe studies involving humans were approved by The Ethics Committee of Beijing Children's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin.
Author contributionsJC: Data curation, Writing – original draft, Methodology. LZ: Methodology, Writing – original draft. JY: Methodology, Writing – review & editing. SZ: Investigation, Writing – review & editing. JJ: Data curation, Investigation, Methodology, Writing – review & editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
AcknowledgmentsWe thank the patients, nurses, and administrators for their participation in the study. The authors would also like to acknowledge all doctors in our department for their contributions to the treatment of patients with hereditary spherocytosis.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References2. Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis–2011 update. Br J Haematol. (2012) 156(1):37–49. doi: 10.1111/j.1365-2141.2011.08921.x
PubMed Abstract | Crossref Full Text | Google Scholar
3. Eber S, Lux SE. Hereditary spherocytosis–defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. (2004) 41(2):118–41. doi: 10.1053/j.seminhematol.2004.01.002
PubMed Abstract | Crossref Full Text | Google Scholar
5. Aggarwal A, Jamwal M, Sharma P, Sachdeva MU, Bansal D, Malhotra P, et al. Deciphering molecular heterogeneity of Indian families with hereditary spherocytosis using targeted next-generation sequencing: first South Asian study. Br J Haematol. (2020) 188(5):784–95. doi: 10.1111/bjh.16244
PubMed Abstract | Crossref Full Text | Google Scholar
6. Tole S, Dhir P, Pugi J, Drury LJ, Butchart S, Fantauzzi M, et al. Genotype-phenotype correlation in children with hereditary spherocytosis. Br J Haematol. (2020) 191(3):486–96. doi: 10.1111/bjh.16750
PubMed Abstract | Crossref Full Text | Google Scholar
7. van Vuren A, van der Zwaag B, Huisjes R, Lak N, Bierings M, Gerritsen E, et al. The complexity of genotype-phenotype correlations in hereditary spherocytosis: a cohort of 95 patients: genotype-phenotype correlation in hereditary spherocytosis. Hemasphere. (2019) 3(4):e276. doi: 10.1097/HS9.0000000000000276
PubMed Abstract | Crossref Full Text | Google Scholar
8. Wu C, Xiong T, Xu Z, Zhan C, Chen F, Ye Y, et al. Preliminary study on the clinical and genetic characteristics of hereditary spherocytosis in 15 Chinese children. Front Genet. (2021) 12:652376. doi: 10.3389/fgene.2021.652376
PubMed Abstract | Crossref Full Text | Google Scholar
9. Kang M, Li H, Zhu J, Zhu L, Hong Y, Fang Y. Clinical manifestations of 17 Chinese children with hereditary spherocytosis caused by novel mutations of the ANK1 gene and phenotypic analysis. Front Genet. (2023) 14:1088985. doi: 10.3389/fgene.2023.1088985
PubMed Abstract | Crossref Full Text | Google Scholar
10. Wang C, Cui Y, Li Y, Liu X, Han J. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable Rare Dis Res. (2015) 4(2):76–81. doi: 10.5582/irdr.2015.01002
PubMed Abstract | Crossref Full Text | Google Scholar
13. Kingdom R, Wright CF. Incomplete penetrance and variable expressivity: from clinical studies to population cohorts. Front Genet. (2022) 13:920390. doi: 10.3389/fgene.2022.920390
PubMed Abstract | Crossref Full Text | Google Scholar
14. Bailey JW, Williams J, Bain BJ, Parker-Williams J, Chiodini PL, General Haematology Task Force of the British Committee for Standards in Haematology. Guideline: the laboratory diagnosis of malaria. Br J Haematol. (2013) 163(5):573–80. doi: 10.1111/bjh.12572
PubMed Abstract | Crossref Full Text | Google Scholar
15. King MJ, Garçon L, Hoyer JD, Iolascon A, Picard V, Stewart G, et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. (2015) 37(3):304–25. doi: 10.1111/ijlh.12335
PubMed Abstract | Crossref Full Text | Google Scholar
16. Hunt L, Greenwood D, Heimpel H, Noel N, Whiteway A, King MJ. Toward the harmonization of result presentation for the eosin-5'-maleimide binding test in the diagnosis of hereditary spherocytosis. Cytometry B Clin Cytom. (2015) 88(1):50–7. doi: 10.1002/cytob.21187
PubMed Abstract | Crossref Full Text | Google Scholar
17. King MJ, Behrens J, Rogers C, Flynn C, Greenwood D, Chambers K. Rapid flow cytometric test for the diagnosis of membrane cytoskeleton-associated haemolytic anaemia. Br J Haematol. (2000) 111(3):924–33.11122157
PubMed Abstract | Google Scholar
18. He BJ, Liao L, Deng ZF, Tao YF, Xu YC, Lin FQ. Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta Haematol. (2018) 139(1):60–6. doi: 10.1159/000486229
PubMed Abstract | Crossref Full Text | Google Scholar
20. Yawata Y, Kanzaki A, Yawata A, Doerfler W, Ozcan R, Eber SW. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol. (2000) 71(2):118–35.10745622
PubMed Abstract | Google Scholar
21. Yang L, Shu H, Zhou M, Gong Y. Literature review on genotype-phenotype correlation in patients with hereditary spherocytosis. Clin Genet. (2022) 102(6):474–82. doi: 10.1111/cge.14223
PubMed Abstract | Crossref Full Text | Google Scholar
22. Park J, Jeong DC, Yoo J, Jang W, Chae H, Kim J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin Genet. (2016) 90(1):69–78. doi: 10.1111/cge.12749
PubMed Abstract | Crossref Full Text | Google Scholar
23. Wong EY, Lin J, Forget BG, Bodine DM, Gallagher PG. Sequences downstream of the erythroid promoter are required for high level expression of the human alpha-spectrin gene. J Biol Chem. (2004) 279(53):55024–33. doi: 10.1074/jbc.M408886200
PubMed Abstract | Crossref Full Text | Google Scholar
24. Delaunay J, Nouyrigat V, Proust A, Schischmanoff PO, Cynober T, Yvart J, et al. Different impacts of alleles alphaLEPRA and alphaLELY as assessed versus a novel, virtually null allele of the SPTA1 gene in trans. Br J Haematol. (2004) 127(1):118–22. doi: 10.1111/j.1365-2141.2004.05160.x
PubMed Abstract | Crossref Full Text | Google Scholar
25. Randon J, Boulanger L, Marechal J, Garbarz M, Vallier A, Ribeiro L, et al. A variant of spectrin low-expression allele alpha LELY carrying a hereditary elliptocytosis mutation in codon 28. Br J Haematol. (1994) 88(3):534–40. doi: 10.1111/j.1365-2141.1994.tb05070.x
PubMed Abstract | Crossref Full Text | Google Scholar
26. Chonat S, Risinger M, Sakthivel H, Niss O, Rothman JA, Hsieh L, et al. The spectrum of SPTA1-associated hereditary spherocytosis. Front Physiol. (2019) 10:815. doi: 10.3389/fphys.2019.00815
PubMed Abstract | Crossref Full Text | Google Scholar
27. Wichterle H, Hanspal M, Palek J, Jarolim P. Combination of two mutant alpha spectrin alleles underlies a severe spherocytic hemolytic anemia. J Clin Invest. (1996) 98(10):2300–7. doi: 10.1172/JCI119041
PubMed Abstract | Crossref Full Text | Google Scholar
28. Peters LL, Jindel HK, Gwynn B, Korsgren C, John KM, Lux SE, et al. Mild spherocytosis and altered red cell ion transport in protein 4.2-null mice. J Clin Invest. (1999) 103(11):1527–37. doi: 10.1172/JCI5766
PubMed Abstract | Crossref Full Text | Google Scholar
29. Kizhatil K, Yoon W, Mohler PJ, Davis LH, Hoffman JA, Bennett V. Ankyrin-G and beta2-spectrin collaborate in biogenesis of lateral membrane of human bronchial epithelial cells. J Biol Chem. (2007) 282(3):2029–37. doi: 10.1074/jbc.M608921200
PubMed Abstract | Crossref Full Text | Google Scholar
30. Peng GX, Yang WR, Zhao X, Jin LP, Zhang L, Zhou K, et al. The characteristic of hereditary spherocytosis related gene mutation in 37 Chinese hereditary spherocytisis patients. Zhonghua Xue Ye Xue Za Zhi. (2018) 39(11):898–903. doi: 10.3760/cma.j.issn.0253-2727.2018.11.005
留言 (0)