
記住我
Neurodegeneration causes an irreversible decline in cognition and motor coordination due to the progressive breakdown of neuronal structure and function. A defining feature of neurodegeneration is the accumulation of misfolded protein aggregates, which are toxic to the cell and cause neuronal damage by disrupting essential cellular processes. A fundamental mechanism in the formation of these aggregates is disruption of neuronal proteostasis, the balance of protein synthesis and degradation. This is mediated in part through the proteasome, a multi-subunit protease complex responsible for the majority of protein degradation, including the misfolded and damaged proteins implicated in neurodegenerative diseases (Zheng et al., 2016; Türker et al., 2021; Cuanalo-Contreras et al., 2023; Davidson and Pickering, 2023). The proteasome functions through multiple proteolytic mechanisms based on its composition and interactors.
The cell’s main degradative machinery, called the 26S proteasome, consists of a cylindrical 20S core particle (20S) that contains catalytic sites for proteolysis and a 19S regulatory cap (19S) that acts as a proteasome activator (PA) to facilitate recognition, unfolding, and rapid degradation of substrates. The 26S proteasome is the central hub for the ubiquitin-proteasome system (UPS), a catabolic pathway that targets proteins for destruction through 1) covalent attachment of polyubiquitin chains by a series of ubiquitin ligases, 2) recognition and de-ubiquitination by the 19S cap, and 3) ATPase-dependent unfolding and translocation of substrate proteins to the interior of the 20S core for degradation (Hershko et al., 1981; Hershko and Ciechanover, 1992; Ciechanover and Schwartz, 1998). The catalytic subunits of the 20S core include β5 (PSMB5; chymotrypsin-like activity), β2 (PSMB7; trypsin-like activity), and β1 (PSMB6; caspase-like activity), which cleave peptide bonds with different specificities and are responsible for the breakdown of proteins into short peptides (Baumeister et al., 1997; Kisselev et al., 1999; Unno et al., 2002). These peptide products are then used as a source of amino acids for biosynthesis or for other cell type-specific functions including antigen recognition, modulation of neuronal signaling, and intercellular communication (Vabulas and Hartl, 2005; Basler et al., 2013; Ramachandran and Margolis, 2017; Limanaqi et al., 2019; Türker et al., 2024). While the UPS is the best-characterized mechanism of proteasome activity (Bingol and Schuman, 2005; Patrick, 2006; Yi and Ehlers, 2007) and extensive reviews have been written on its role in neurodegenerative diseases (Ciechanover and Brundin, 2003; Dantuma and Bott, 2014; Zheng et al., 2016; Watanabe et al., 2020; Schmidt et al., 2021; Davidson and Pickering, 2023), many questions remain which are being actively explored and which will not be the focus of this review.
In addition to the UPS, the 20S proteasome is highly abundant and found with a broad set of activators and associated proteins important for various proteolytic functions, especially those affected by neurodegenerative disease (Fabre et al., 2014; Opoku-Nsiah and Gestwicki, 2018; Türker et al., 2023). While 20S proteasomes were previously believed to be non-functional without a regulatory 19S cap, increasing evidence has indicated unique, ubiquitin-independent roles of the 20S core particle and its interacting partners, particularly in degradation of intrinsically disordered, oxidized, or misfolded proteins (Jariel-Encontre et al., 2008; Baugh et al., 2009; Ben-Nissan and Sharon, 2014; Erales and Coffino, 2014; Opoku-Nsiah and Gestwicki, 2018; Davidson and Pickering, 2023), important hallmarks of neurodegeneration. Because a large portion of proteins in the human genome contain intrinsically disordered regions under physiological conditions, and because 20% of proteins may be degraded through ubiquitin-independent proteasome pathways under normal or stress conditions, it is likely that these pathways are more important for quotidian function than previously appreciated (Baugh et al., 2009; Ben-Nissan and Sharon, 2014; Pepelnjak et al., 2024). In this review, we focus on ubiquitin-independent proteasomal mechanisms and the emerging role these mechanisms plays in neurodegeneration.
Search scheme and article selectionPubMed and Google Scholar search engines were first used to identify research articles using search terms including ubiquitin-independent proteasome, neurodegeneration, 20S proteasome, Alzheimer’s Disease, Parkinson’s Disease, Huntington’s Disease, protein aggregation, oxidative stress, PA200, PA28, and aging, among others. However, results from these keyword search terms did not distinguish well between the UPS and ubiquitin-independent mechanisms, so alternative tools employing artificial intelligence (AI) were used. These included Consensus and Semantic Scholar–the primary tools used–as well as Elicit and Research Rabbit. Consensus, Elicit, and Semantic Scholar were leveraged to search databases of >200 million peer-reviewed scientific papers using natural language processing to interpret questions about research topics rather than keywords (e.g., “What roles do ubiquitin-independent proteasome mechanisms play in neurodegenerative disease?” or “Can the 20S proteasome degrade tau without a proteasome activator?”), using machine learning to process the content and context of literature, and using large language models to suggest relevant articles or provide a summary of conflicts and consensus in the literature with references, reducing bias in the search results. After curating a collection of the most relevant articles based on these searches, Research Rabbit was used to visualize connected papers, identifying additional article suggestions. Aside from AI tools, other papers were identified by scanning the references of pertinent articles. After identification, full-text articles published in or before October 2024 were reviewed for ubiquitin-independent proteasome mechanistic relevance. Original articles and reviews were included, and retracted papers were excluded. All articles were peer-reviewed except for one pre-print (indicated in the text). The writing of this review was not AI-generated, and AI tools were used for article identification only.
Proteasome substrate identificationFor decades, evidence has demonstrated that the proteasome has diverse mechanisms of substrate recognition and degradation beyond the canonical UPS pathway (Jariel-Encontre et al., 2008; Baugh et al., 2009; Ben-Nissan and Sharon, 2014; Erales and Coffino, 2014), and a growing number of protein substrates targeted to the proteasome without ubiquitination have been discovered (Rosenberg-Hasson et al., 1989; Bercovich and Kahana, 1993; Coffino, 1998; Sheaff et al., 2000; Bossis et al., 2003; Myers et al., 2018; Makaros et al., 2023). Recent advances include a systematic analysis of human 20S proteasome substrates using a method called proteasomal-induced proteolysis mass spectrometry, developed by Pepelnjak et al. to identify a range of proteins degraded by the ubiquitin-independent 20S (Pepelnjak et al., 2024). Another technique, Global Protein Stability peptidome screening, was developed by Koren et al. (2018) and subsequently applied to identify ubiquitin-independent proteasome substrates (Makaros et al., 2023). These papers and others have demonstrated that proteins central to neurodegeneration, including tau (important in AD and other tauopathies) (David et al., 2002; Grune et al., 2010; Ukmar-Godec et al., 2020), α-synuclein (important in PD and other synucleinopathies) (Tofaris et al., 2001; Nakajima et al., 2005; Alvarez-Castelao et al., 2014; Makaros et al., 2023), huntingtin (important in HD) (Juenemann et al., 2013), as well as many proteins important in stress, transcriptional regulation (like RNA-binding partners and transcription factors), phase granule separation (Myers et al., 2018), and cell cycle regulation (Sheaff et al., 2000; Touitou et al., 2001; Asher et al., 2005; Wiggins et al., 2011), can be degraded through ubiquitin-independent mechanisms (Pepelnjak et al., 2024). These techniques suggest that ubiquitin-independent degradation is far more prevalent than previously believed, although in some cases, more orthogonal approaches or in vivo data approximating normal physiology may be needed to definitively support this claim. Although significant advances are rapidly emerging, the exact targeting mechanisms of ubiquitin-independent degradation are still under investigation. However, the 20S proteasome is known to degrade intrinsically disordered proteins (IDPs) like α-synuclein (α-syn) and tau more efficiently than structured proteins (Grune et al., 2010; Alvarez-Castelao et al., 2014; Myers et al., 2018; Ukmar-Godec et al., 2020), and there may be additional specific motifs (Touitou et al., 2001; Bossis et al., 2003; Makaros et al., 2023), including C-terminal degrons (Touitou et al., 2001; Makaros et al., 2023), and structural features including exposed hydrophobic residues (Kisselev et al., 2002), that are recognized for targeted degradation. It has also recently been demonstrated that the 20S can degrade ubiquitinated substrates, degrading the ubiquitin tag along with the protein more quickly than the 26S can deubiquitinate and digest, a mechanism increased during hypoxic stress conditions to clear misfolded/damaged proteins rapidly (Sahu et al., 2021).
To protect the cell from excessive proteolysis, entry into the catalytic chamber of the 20S core is tightly regulated, with its external α-rings partially obstructing the protease active sites (Wenzel and Baumeister, 1995; Groll et al., 2000). To allow substrate entry, 20S proteasomes may interact with pore-opening proteasome activators (PAs) (Knowlton et al., 1997; Hendil et al., 1998; Ortega et al., 2005), or they may allow direct substrate access without a PA (Kisselev et al., 2002). As a standalone molecule, the 20S can recognize and interact with hydrophobic regions of misfolded proteins, which act as degradation signals, as well as IDPs and oxidized proteins (Kisselev et al., 2002; Förster et al., 2003; Raynes et al., 2016; Deshmukh et al., 2023). Because IDPs lack a rigid, well-defined structure, they are more flexible and can more easily enter the narrow entry channel of the 20S proteasome (Suskiewicz et al., 2011), whereas structured proteins require unfolding or linearization by the 19S cap ATPases (Wenzel and Baumeister, 1995; Dong et al., 2019). The independent 20S can undergo conformational changes in its α-rings without ATP hydrolysis that permit self-gated entry of unstructured, oxidized, or misfolded proteins through the narrow entry pore (Kisselev et al., 2002; Förster et al., 2003). This capacity for protein degradation without ubiquitination or energy consumption makes the 20S proteasome uniquely suited to remediate the accumulation of toxic protein aggregates in neurodegenerative diseases, which often cause mitochondrial damage, oxidative stress, and further impairment of the UPS (Ding et al., 2006; Pickering et al., 2010; Li et al., 2011; Huang et al., 2013; Höhn et al., 2020). Importantly, substrate degradation by ubiquitin-independent proteasomal mechanisms, the UPS, or non-proteasomal pathways like autophagy are not necessarily mutually exclusive in the cell, and some substrates may be degraded by one mechanism in some conditions and another mechanism in other conditions, such as oxidative stress (Grune et al., 2010; Suskiewicz et al., 2011; Ben-Nissan and Sharon, 2014; Manfredonia and Kraut, 2022). It is also well-documented that autophagic pathways may be used to clear certain isoforms of these proteins, hypermodified forms, or aggregates, which will not be discussed here (Lee et al., 2013; Watanabe et al., 2020). Increasing research has shed light on how these substrates are targeted and the variety of mechanisms used to facilitate or regulate their degradation through the proteasome.
Proteasome activators (PAS)The best-understood mechanisms of substrate targeting are through PAs. Prior research has demonstrated that interaction with 20S molecules require many PAs to use a C-terminal tri-peptide HbYX motif (hydrophobic residue, followed by tyrosine, then any amino acid) that docks into the spaces between ⍺-ring subunits, called 20S ⍺-pockets (Smith et al., 2007; Rabl et al., 2008; Sadre-Bazzaz et al., 2010). In contrast to research on the HbYX motif in archaea models (Smith et al., 2007; Rabl et al., 2008; Yu et al., 2010), recent research shows that human 20S proteasomes, which are hetero-oligomers with seven distinct ⍺-subunits in the outer ⍺-ring rather than the homo-oligomers formed by archaea, may have more heterologous signals, dubbed YΦ motifs by the Gestwicki group in 2022 (Opoku-Nsiah et al., 2022). While HbYX motifs are tripeptides, the YΦ motifs tested were hexapeptide sequences, showing an effect on degradation for each of the last 6 residues of the C-terminus. YΦ refers to Y-F/Y residues at the antepenultimate and penultimate positions at the C-terminus. In addition, as opposed to monovalent PAs like PA200 that require adherence to HbYX/YΦ rules (Sadre-Bazzaz et al., 2010), hetero-oligomeric PAs, which have increased valency due to interactions with multiple ⍺-subunit pockets, allowed some flexibility in C-terminal gating association outside of HbYX/YΦ rules (Opoku-Nsiah et al., 2022). Further research must be performed to catalog the full range of interaction sequences present in human 20S proteasomes, and this may provide insight into regulation of PAs, the importance of any post-translational modifications, and their role in neurodegenerative diseases.
An important implication of these recognition sequences is that they may be useful as drug targets. It has been demonstrated that synthesized HbYX-like peptide mimetics can open the 20S pore and stimulate degradation of unstructured protein substrates, posing a possible therapeutic option for neurodegenerative disease (Chuah et al., 2023). In addition to increasing degradation of tau by the 20S, HbYX mimetics also completely block 20S inhibition by amyloid-β, α-syn, and huntingtin oligomers, further demonstrating the potential for small molecule treatments to restore 20S proteasome activity and increase degradation of disordered substrates prone to aggregation (Chuah et al., 2023).
The most famous PA is the 19S regulatory cap (also called PA700), which in addition to being the canonical activator complex in the UPS has some capacity for facilitating ubiquitin-independent degradation through the 26S proteasome, generating a different set of peptides than 20S alone (Kisselev et al., 1999; Asher et al., 2005; Baugh et al., 2009; Winkler et al., 2013; Ben-Nissan and Sharon, 2014; Tsvetkov et al., 2020). In addition, Tsvetkov et al. showed in vitro that when the assembled 26S is stabilized by binding of NADH, an important molecule in aging and metabolism that is sensitive to cellular redox state, it can facilitate degradation of IDPs even in the absence of ATP. However, 20S proteasome catalytic activity was not affected by NADH or NAD+ in vitro (Tsvetkov et al., 2020). Other PAs that can bind to the exterior of the 20S core and enhance its activity by inducing central pore opening include PA28 (also called 11S or PSME1) and PA200 (or Blm10 in yeast), both of which use ubiquitin-independent mechanisms to facilitate substrate entry and often target oxidized, unstructured/intrinsically-disordered, or misfolded proteins in specific subcellular locations (Dubiel et al., 1992; Ma et al., 1992; Ustrell et al., 2002; Ortega et al., 2005; Cascio, 2021). In addition to PAs, other major proteasome interactors include PI31 (also called PSMF1), an adaptor protein for proteasome transport in neurons (Liu et al., 2019); midnolin, a regulator of immediate early gene protein and transcription factor degradation (Gu et al., 2023); ECPAS (“Ecm29 proteasome adaptor and scaffold”; also called PSMG1, or Ecm29), a modulator of 26S assembly and disassembly participating in stress responses (Wang et al., 2017; Lee et al., 2020), and catalytic core regulators (CCRs), which allosterically modulate uncapped 20S activity (Deshmukh et al., 2023). These will be explored in greater detail in the following subsections. See Figure 1 and Table 1.
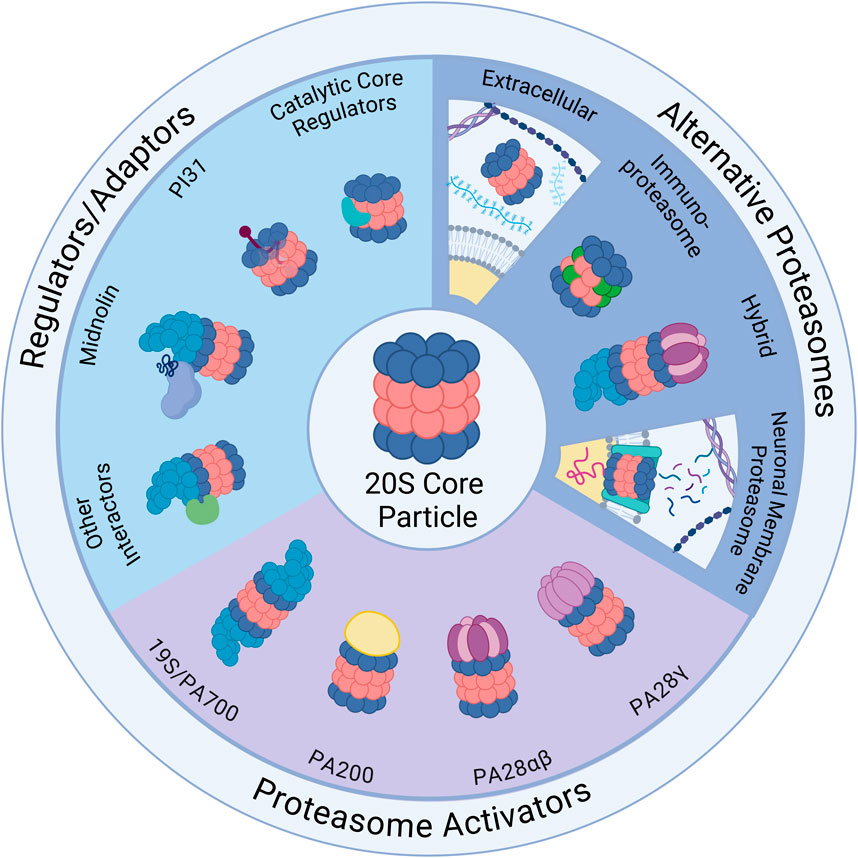
Figure 1. Ubiquitin-Independent Proteasome Complexes and Mechanisms. Illustrated above are the diverse complexes that can form in combination with the 20S core particle (20S) (center illustration). The 20S has 28 subunits that form a barrel structure with 14 α-subunits (blue) and 14 β-subunits (red). Alternative Proteasomes: The 20S can be found in various forms including the immunoproteasome, which has alternative β catalytic subunits (green) and can be induced as part of the immune response, and the neuronal membrane proteasome (NMP), which is a neuron-specific proteasome complex localized to the plasma membrane that is used for signaling. The 20S can also be found in the extracellular space and can exist in hybrid forms which have two distinct cap structures on each side of the 20S (shown here with 19S and PA28αβ). Regulators/Adaptors: The 20S interacts with several important regulators/adaptors including catalytic core regulators (CCRs), midnolin, PI31, and a broad category encompassing other interactors. Aside from PI31, which interacts with 20S subunits from inside the barrel, most of these regulators associate with the 20S exterior, and in some cases (midnolin and several other interactors) with the 19S-capped 20S. Proteasome Activators (PAs): The 20S interacts with a variety of PAs that increase 20S activity by opening the gate formed by α-subunits and permitting substrate entry for degradation. Those pictured include: the 19S cap, which combines with the 20S to form the 26S; PA200, which is monomeric and mainly found in the nucleus; PA28αβ, which is typically cytoplasmic and plays an important role in the immune response; and PA28γ, which has high expression in the brain and serves an important role in cell cycle regulation. Created in BioRender. Church, T. (2025) https://BioRender.com/o73i883.
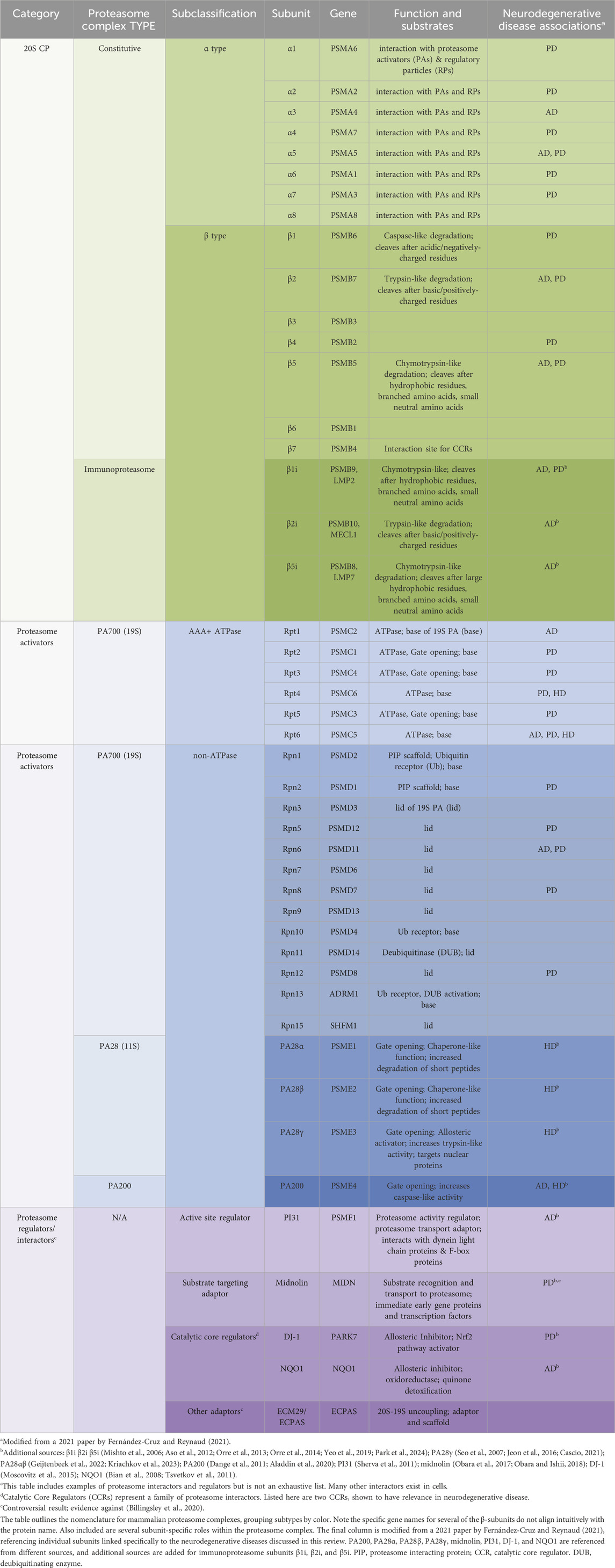
Table 1. Proteasome nomenclature and subunit-specific links to neurodegenerative disease.
PA28PA28 is a heptameric, ring-shaped, and ATP- and ubiquitin-independent 20S PA that promotes rapid degradation of small, unstructured protein fragments, short peptides, and oxidized or misfolded proteins in the nucleus and cytoplasm by binding to the ends of the 20S core and inducing conformational changes that widen the 20S pore (Ma et al., 1992; Knowlton et al., 1997; Zhang et al., 1999; Thomas and Smith, 2022). Importantly, there are multiple isoforms of PA28, including PA28α (also called REGα and PSME1), PA28β (also called REGβ and PSME2), and PA28γ (also called REGγ and PSME3), which have distinct sets of functions and substrates (Knowlton et al., 1997; Thomas and Smith, 2022). PA28α and PA28β are mainly cytoplasmic and typically combine to form heteroheptamers, but PA28α is expressed at higher levels in the brain than PA28β and can form a homoheptamer (Knowlton et al., 1997; Zhang et al., 1999; Noda et al., 2000). PA28γ also forms homoheptamers and is primarily nuclear, ubiquitously expressed in all organ systems, with particularly high expression in the brain (Noda et al., 2000; Cascio, 2021; Frayssinhes et al., 2021). PA28γ is an interferon-γ- (IFNγ) and ubiquitin-independent PA which serves as a regulator of DNA replication, DNA repair, transcription, cell cycle control, and p53 tumor suppressor stability (Zhang and Zhang, 2008).
Because it is not an ATPase, PA28 cannot unfold proteins and has a preference for disordered or partially unfolded proteins, which can enter the 20S catalytic chamber without additional unfolding (Frayssinhes et al., 2021). It can interact with both the typical 20S core (constitutive 20S) and the immunoproteasome (a modified, inducible complex described in the “Immunoproteasome” section of this review), inducing different allosteric effects on each (Lesne et al., 2020). Indeed, PA28 becomes increasingly important in aging and neurodegenerative disease, when the UPS is compromised and damaged proteins accumulate (Seo et al., 2007), through its regulation of 20S activity, its ability to activate 26S as a hybrid proteasome (19S-20S-11S/PA28) (Tanahashi et al., 2000), and as a standalone chaperone-like molecule and chaperone regulator (Minami et al., 2000; Adelöf et al., 2018; Adelöf et al., 2021). PA28 expression is also upregulated under conditions of high protein damage including oxidative stress, indicating an important role in maintaining proteostasis by mitigating oxidative damage, and it often accompanies upregulation of the immunoproteasome (Pickering et al., 2010; Pickering et al., 2012).
While best characterized in other cell types, studies in neurons have shown that PA28 promotes ubiquitin-independent proteasomal degradation of oxidized and misfolded proteins and protects against oxidative stress (Li et al., 2011; Pickering and Davies, 2012), increasing evidence for its role as a 20S regulator in oxidatively-burdened neurodegenerative disease states. In addition, PA28⍺β overexpression showed sex-specific benefits for female mice in preventing age-related protein aggregation, hypothesized by the authors to be a novel, proteasome-independent, chaperone-like function (Adelöf et al., 2018). Moreover, PA28⍺β, plays major roles in the immune system through regulation of the immunoproteasome, described in the “Immunoproteasome” section below. In neurons and microglia, exposure to cytokine IFNγ or other pro-inflammatory factors during an immune or inflammatory response increases PA28αβ expression and its association with the immunoproteasome (Rivett et al., 2001; Pickering et al., 2010; Pintado et al., 2012). Because neuroinflammation is increasingly recognized as a contributor to the development of neurodegenerative diseases, dysregulation of PA28αβ - and therefore the immunoproteasome - can contribute to neuroinflammatory processes through neurons and glia (Leng and Edison, 2021; Malek et al., 2024). Notably, studies have also reported altered PA28 expression in Alzheimer’s disease brains (Krzyzanowska et al., 2015) and PA28γ may play a complex role in the etiology of HD, which will be described in the “Huntington’s Disease” section below (Cascio, 2021). Expansion on the significance of PA28 in specific neurodegenerative disease will be included in sections below.
PA200PA200 is a large, monomeric, and ATP- and ubiquitin-independent 20S proteasome activator found predominantly in the nucleus which associates with the 20S core and regulates DNA repair mechanisms, transcription, and the cell cycle through targeted, acetylation-dependent degradation of histones and other protein targets (Ustrell et al., 2002). Its structure has two apertures for substrate entry and forms a dome-like cap on the 20S to open it (Guan et al., 2020). Some evidence suggests PA200 alters the relative activity of the 20S β catalytic subunits, increasing β1 (Ustrell et al., 2002) or β2 (Toste Rêgo and Da Fonseca, 2019) activity compared to the uncapped 20S.
Because PA200 does not have ATPase activity, it primarily acts on peptides and disordered and partially unfolded proteins, although there is a possibility it has some intrinsic unfolding ability through recruitment of other factors or conformational changes of substrates. In addition to possible regulatory roles in proteasome stability or maturation (VerPlank et al., 2024), PA200 is upregulated in response to DNA damage and induces opening of the α-ring substrate entry channel of the 20S, allowing for rapid clearance of oxidized, aggregated, and misfolded substrates (Ortega et al., 2005). These substrates include tau (Dange et al., 2011) and N-terminal huntingtin protein fragments (Aladdin et al., 2020), the proteins responsible for the pathogenic aggregates in AD and HD, respectively. Because research on PA200 function in the nervous system is still limited, its role in neurodegenerative diseases, its regulation and interactions with other PAs, and its cell type-specific characteristics in neurons remain mostly unknown. In fact, depending on the disease state, PA200 may ameliorate or worsen neurodegeneration in in vivo disease models (Aladdin et al., 2020; VerPlank et al., 2024).
Additional cap conformations and hybrid proteasomesFurther research is being performed to investigate the existence and roles of additional alternative caps, including hybrid proteasomes with different caps (e.g., one 19S and one alternative cap associated with a 20S core) (Hendil et al., 1998; Tanahashi et al., 2000; Cascio et al., 2002). While initially thought to be absent from the brain (Noda et al., 2000), along with PA28β, more recent data have demonstrated the presence of hybrid proteasomes and PA28β (Adelöf et al., 2018; Kriachkov et al., 2023). The significance of hybrid proteasomes in neurons is not yet well understood, but they may provide finely calibrated regulation of substrates or alter proteasome catalytic activity to generate a different set of peptides, as has been seen in the immune system to modify peptide products for antigen presentation (Hendil et al., 1998; Cascio et al., 2002). Hybrid proteasomes have also been proposed to use the 19S for protein unfolding and entry into the 20S chamber and PA28 for the rapid release of digestion products (Pratt and Rechsteiner, 2008; Kriachkov et al., 2023). It is unknown if PI31 or other adaptors are involved in hybrid proteasomes.
In addition, there may be tissue-specific, ubiquitin-independent alternative caps, as has been noted for ubiquitin-dependent PAs (Goldberg et al., 2021), or transient caps that do not assemble into stable structures like PA28 or PA200 but which interact briefly with the 20S core to modulate its activity or substrate channel access (Clemen et al., 2015; Esaki et al., 2018; Goldberg et al., 2021). These phenomena may be especially likely in neurons, which have specialized proteasomes, unique activators, and additional proteasomal physiological functions (Bingol and Schuman, 2005; Ramachandran and Margolis, 2017; Goldberg et al., 2021). An example of this is the characterization of neurodegenerative disease associated protein valosin-containing protein (also called Cdc48, TER94, and p97), a HbYX motif-containing PA which plays a role in ubiquitin-dependent degradation (Johnson et al., 2010; Esaki et al., 2018). In addition, some evidence suggests that 19S caps may not require ubiquitin for degradation of some substrates (Kisselev et al., 1999; Winkler et al., 2013; Tsvetkov et al., 2020). The full range of endogenous proteasome interactors and alternative caps are still being explored, and more molecules are likely to emerge.
Proteasome regulatorsIn addition to PAs, other proteasome adaptor and interactor proteins can regulate proteasome assembly and disassembly, link the proteasome to signaling pathways, regulate substrate specificity, and direct intracellular trafficking of proteasomes. The functions of these adaptors vary by cellular conditions, cell type, and activation of intersecting regulatory pathways (Arkinson et al., 2024). See Figure 1 and Table 1.
PI31While not a PA, PI31 has been proposed as an endogenous 20S proteasome regulator and is targeted to the 20S via a HbYX motif. In vitro, it interacts with both 20S and 26S constitutive proteasomes and has been found to inhibit the 20S and to prevent binding of the 19S cap and PA28 (McCutchen-Maloney et al., 2000; Li et al., 2014; Liu et al., 2019; Wang et al., 2024). In contrast to the constitutive 20S and 26S, immunoproteasomes are capable of cleaving the PI31 C-terminus, preventing its binding and its inhibition of the core catalytic subunits (Wang et al., 2024). Separately, another study suggests PI31 affects immunoproteasome assembly (Zaiss et al., 2002). Although much of the research regarding PI31 has been performed in vitro (Li et al., 2014; Wang et al., 2024), in vivo experiments have contributed to a complex picture of PI31-mediated proteasome regulation. In a more physiological context, PI31 may in fact activate proteasome degradation through the 26S, and in addition, both knockout and overexpression of PI31 are lethal, indicating that cells are sensitive to PI31 amount (Bader et al., 2011). In vivo experiments in mouse motor neurons have demonstrated that PI31 acts as a proteasome regulator and adaptor protein that connects the proteasome to transport machinery for translocation down neuronal projections including axons, an essential function for maintaining a healthy proteasome supply to diverse cellular locations for their various functions (Bader et al., 2011; Liu et al., 2019). Loss of PI31 contributes to neurodegeneration, as its regulatory activity is required for normal proteasome function, maintenance of synapses, and neuronal survival (Bader et al., 2011; Liu et al., 2019; Minis et al., 2019). Genome-wide association studies have linked PI31 to AD risk (Sherva et al., 2011), and a direct antagonist of PI31, called valosin-containing protein (VCP), causes a familial type of the neurodegenerative disorder amyotrophic lateral sclerosis (Johnson et al., 2010; Clemen et al., 2015).
Catalytic core regulators (CCRs)As the importance of non-UPS proteasome activity is becoming more apparent, it is increasingly critical to study regulators of these mechanisms. A newly discovered family of multi-functional regulatory proteins that directly interact with the 20S core to closely modulate its cap-independent degradation of IDPs and damaged, partially-unfolded proteins are the Catalytic Core Regulators (CCRs) (Olshina et al., 2020; Deshmukh et al., 2023). These CCRs are allosteric regulators with shared structural features including a common N-terminal sequence motif and a Rossman fold, providing further evidence that HbYX motifs represent only a portion of the structural features characterizing proteasome regulators. CCRs bind to the external surface of the 20S β7 (PSMB4) subunit and induce a conformational change that inhibits all three catalytic mechanisms of degradation without plugging the substrate entry gate and can protect substrates from degradation, including ⍺-syn, which forms toxic oligomers in PD and other synucleinopathies (Deshmukh et al., 2023). CCRs are critical for coordinating the oxidative stress response through interaction with transcription factor Nrf2 and activation of a range of response factors including upregulation of 20S subunits (Olshina et al., 2020; Deshmukh et al., 2023). The identification of the structural features underlying allosteric regulation of degradation by the 20S also provides insight relevant to the development of selective, synthesized inhibitors of 20S proteasomes and possible therapeutic options for neurodegenerative diseases like PD, which is directly affected by a CCR called DJ-1 and is discussed later in this review (Moscovitz et al., 2015).
MidnolinMidnolin is an inducible, chaperone-like protein that associates with the 26S proteasome to promote selective, ubiquitin-independent degradation of transcription factors and other nuclear proteins (Gu et al., 2023). Recent studies indicate its targeting mechanism uses an internally symmetrical “catch” domain that induces a conformational change in unstructured regions of protein substrates to capture them for destruction by the proteasome. Midnolin associates with the proteasome using a C-terminal ⍺ helix, which does not contain a HbYX motif or YΦ motif, through an unknown mechanism, and it facilitates degradation of targets bound to the catch domain using an N-terminal ubiquitin-like domain. While the reasons for its preference for the 26S over the 20S are not well understood, midnolin co-immunoprecipitates with both 19S and 20S subunits (Gu et al., 2023).
Substrates of midnolin include transcriptional regulators and immediate early gene products, which are rapidly induced upon stimulation by a variety of stimuli and regulate transcription of longer-term sets of proteins in response to a particular stimulus (Chiba et al., 2024; Gu et al., 2023). Notably, midnolin and some of its substrates have been identified as PD risk genes, with deletion of midnolin resulting in loss of parkin expression, increased expression of ⍺-syn, and induction of PD phenotypes including loss of neurite outgrowth (Obara et al., 2017; Obara and Ishii, 2018). Obara et al. used microarray analysis to show that 10.5% of sporadic PD patients and 0% of healthy controls lack one copy of midnolin, positing a role for midnolin loss in development of PD (Obara et al., 2017). These data were supported by another large cohort study by the same research team in 2019, which showed a significant odds ratio of 4.35 with midnolin copy number loss for development of PD, with the odds ratio increasing to 22.3 when copy number loss is defined by large deletions (Obara et al., 2019). However, using whole genome sequencing and analysis of a public database of structural variants, Billingsley et al. and the International Parkinson’s Genomics Consortium did not identify PD-associated midnolin deletions and disputed the determination of midnolin as a PD risk gene, indicating that further study with orthogonal methods is required to investigate midnolin association with PD and resolve controversy (Billingsley et al., 2020).
Additional regulators/adaptorsBeyond the above regulators, there are other critical 20S interactors and adaptors that affect 20S activity and assembly briefly described here. Chaperones PAC1-PAC4 and POMP are crucial for the proper formation and maturation of the proteasome from its constituent monomeric subunits (Hirano et al., 2005; Hirano et al., 2006; Fricke et al., 2007; Le Tallec et al., 2007), and adaptors including ECPAS contribute to 26S assembly and disassembly in vivo (Wang et al., 2017; Choi et al., 2023). The ECPAS-20S interaction regulates the 20S:26S ratio and modulates the balance between ubiquitin-dependent and -independent mechanisms for adaptation to conditions like glucose deprivation or oxidative stress, in which it facilitates disassembly of 26S to 20S to support degradation of oxidized and misfolded substrates (Leggett et al., 2002; Wang et al., 2017; Choi et al., 2023). Interactions among the proteasome, ECPAS, and ankyrin G also regulate critical remodeling of the axon initial segment of neurons, found to have significant structural abnormalities in AD-affected neurons (Lee et al., 2020).
In addition to proteins directly bound to the 20S, there are also substrate-bound proteins critical to its regulation, deemed “nanny” proteins, that protect newly synthesized intrinsically disordered 20S substrates from degradation and allow new intrinsically disordered proteins (IDPs) to mature (Tsvetkov et al., 2009). Potential nanny proteins suggested by Tsvetkov et al., have been linked to a variety of nervous system disorders (Enokido et al., 2010; Kamińska et al., 2024; Yuhan et al., 2024). Conversely, chaperones like Hsp70 and Hsp110, dysfunction of which has been linked to neurodegenerative diseases, facilitate the targeting of substrates to the proteasome for ubiquitin-dependent and ubiquitin-independent degradation (Eroglu et al., 2010; Turturici et al., 2011; Hjerpe et al., 2016; Kandasamy and Andréasson, 2018; Taguchi et al., 2019; Vinokurov et al., 2024). Hsp70 has further been demonstrated to interact with ubiquilin2, a shuttling factor that brings substrates to the proteasome and plays a role in neurodegeneration (Wang et al., 2006; Zhang et al., 2014; Hjerpe et al., 2016; Ma et al., 2023). While members of the ubiquilin family typically require ubiquitin for trafficking proteasome degradative targets (Zhang et al., 2014; Itakura et al., 2016), Makaros et al. demonstrated that ubiquilins may also mediate ubiquitin-independent proteasome substrate identification (Makaros et al., 2023).
There are likely many additional yet-uncharacterized proteins that regulate 20S proteasome activity in neurons. In addition, post-translational modifications like phosphorylation, oxidation, or acetylation can also alter proteasome activity and 20S interaction with regulators in various cell types, especially as cells age (Bulteau et al., 2000; Bulteau et al., 2001; Ishii et al., 2005; Kors et al., 2019).
Specialized proteasomesImmunoproteasomeWhen stimulated by interferon-γ (IFNγ) or oxidative stress, immune cells and some other cell types (e.g., microglia in the nervous system (Orre et al., 2013; Malek et al., 2024)) can produce a modified proteasome, the immunoproteasome, which can act through ubiquitin independent or ubiquitin dependent mechanisms and replaces the three catalytic β-subunits (β1, β2, β5) in the constitutive proteasome core with three unique catalytic subunits (β1i, β2i, β5i, also called PSMB9/LMP2, PSMB10/MECL-1, PSMB8/LMP7) (Noda et al., 2000; Basler et al., 2013; Freudenburg et al., 2013; Rock et al., 2014; Johnston-Carey et al., 2015; Ettari et al., 2017; Winter et al., 2017; Abi Habib et al., 2022). This subunit replacement allows for the generation of longer peptides which can be further processed and presented as antigens that allow cells to determine self vs. non-self, an important component of immune responses, although an increasing number of roles for the immunoproteasome are being recognized (Pickering et al., 2010; Abi Habib et al., 2020; Tundo et al., 2023). It appears to be particularly important for the clearance of oxidized and misfolded proteins in response to oxidative stress (Pickering et al., 2010; Pickering and Davies, 2012). Immunoproteasomes are upregulated in reactive glia in AD mouse models (Orre et al., 2013; Orre et al., 2014), which may be critical for elimination of misfolded or damaged proteins that could spread between cells and cause disease progression. Studies using immunoproteasome-specific inhibitors demonstrate improvements in cognitive decline in AD mice, showing that the immunoproteasome may contribute to pathology in association with neurodegeneration (Yeo et al., 2019; Park et al., 2024). The ability to generate highly selective inhibitors for modified immunoproteasome subunits provides a pathway to evaluate the effects of these complexes without affecting constitutive proteasome activity, a therapy that could have benefits in neurodegenerative diseases impacted by neuroinflammation (Johnston-Carey et al., 2015; Malek et al., 2024).
While the expression of immunoproteasome in the young and healthy brain is very low or negligible, and for a long time it was believed that the immunoproteasome was not expressed in the brain at all (Noda et al., 2000), immunoproteasome has been detected in both neurons and glia in aged healthy brains and brains with neurodegeneration (Diaz-Hernandez et al., 2003; Mishto et al., 2006; Aso et al., 2012; Ugras et al., 2018), suggesting that the induction of immunoproteasome in the brain may be a result of aging, neurodegeneration, or neuroinflammation. Neuroinflammation can exacerbate neurodegeneration but requires additional impaired proteasomal degradation to induce disease phenotypes (Pintado et al., 2012; Malek et al., 2024). Indeed, proteasome research in AD brains has demonstrated a notable induction of immunoproteasome subunits and changes in proteasome activity and composition, although the specific changes observed have varied substantially across studies (e.g., a decrease of only trypsin-like activity vs decreases in both chymotrypsin-like and caspase-like activity; a decrease in β1 expression and a proportional increase in β1i/LMP2 expression vs little change in expression levels) (Mishto et al., 2006; Aso et al., 2012; Keller et al., 2000a; Davidson and Pickering, 2023). In contrast to findings showing decreased activity, a recent study using advanced activity-based probes to detect global proteasome activity in human AD brain tissue detected elevated activity (Türker et al., 2023).
An induction of immunoproteasome has also been detected in HD brains and PD brains (Diaz-Hernandez et al., 2003; Ugras et al., 2018). Concurrent with an increase in LMP2 and LMP7 expression, an increase in trypsin- and chymotrypsin-like activity was observed in HD brains in the areas most affected (Diaz-Hernandez et al., 2003), and in PD brains, an increase in expression of immunoproteasome subunit LMP7 (β5i) was observed (Ugras et al., 2018). A recent study in mice found that knocking out immunoproteasome in brain can also cause seizures, tau hyperphosphorylation, increased polyubiquitination, and neurodegeneration, and the authors suggest that immunoproteasome has a role in healthy brain aging (Leister et al., 2024). In-depth descriptions of the immunoproteasome in neurodegeneration can be found in other recent reviews (Zerfas et al., 2020; Tundo et al., 2023).
Neuronal membrane proteasomeA specialized proteasome found in the plasma membrane of neurons, called the neuronal membrane proteasome (NMP), is another form of 20S proteasome that functions through ubiquitin-independent mechanisms (Ramachandran and Margolis, 2017). The NMP degrades nascent polypeptide chains from ribosomes closely associated with the membrane to form its signaling molecules (Ramachandran et al., 2018), but it is not yet known how substrate selection or recognition motifs to the NMP may differ from other 20S proteasomes in neurons. Unlike proteasomes whose primary role is in protein turnover, the NMP degrades intracellular proteins into peptides expelled into the extracellular space, creating small, specific peptide signaling molecules that serve additional functions in neurons that are important in synaptic regulation, including NMDA receptor activity modulation, and in pain sensation modulation (Ramachandran and Margolis, 2017; Ramachandran et al., 2018; Türker et al., 2024; Villalón Landeros et al., 2024). Because the NMP regulates neuronal circuits and is essential in learning-induced behavioral plasticity (He et al., 2023), it is possible that dysfunction of the 20S proteasome induced by aging and proteotoxic aggregates in neurodegeneration may also disrupt NMP function, further contributing to declining cognition in neurodegenerative diseases. Supporting this hypothesis, a preprint in bioRxiv by Paradise et al. showed an association between NMP and ApoE, a critical AD risk gene (Paradise et al., 2023). In their experiments, NMP co-purified with ApoE, suggesting a physical interaction, and inhibition of the NMP was sufficient to cause aggregation of newly-synthesized tau. Future studies will reveal the full details of this novel proteasome complex and its function in brain health and disease.
Extracellular proteasomeWhile proteasomes are typically thought of as intracellular, increasing evidence has shown 20S proteasomes in extracellular vesicles (EVs) and free-floating in a variety of body fluids, including the interstitial fluid and the cerebrospinal fluid of the brain (Mueller et al., 2012; Ben-Nissan et al., 2022). It has been reported that extracellular proteasomes rarely contain 19S or PA200 PAs (Kulichkova et al., 2017; Tsimokha et al., 2020), although this is debated and may be body fluid/tissue specific (Ben-Nissan et al., 2022). However, they are often free floating as 20S or accompanied by PA28αβ or PI31, and they have significant levels of post-translational modifications, including several unique from other proteasome complexes (Tsimokha et al., 2020; Ben-Nissan et al., 2022), which may affect their function or localization to the extracellular space. The mechanisms of extracellular release and the source of these proteasomes are not yet fully understood, but data have shown release of 20S core particles and PA28 molecules by immune cells in microparticles subject to later dissolution (Bochmann et al., 2014; Bonhoure et al., 2022). It is possible that neurons or glia could release proteasomes as part of normal physiology or as part of a stress response (Ben-Nissan et al., 2022), or that cell damage or cell death causes intracellular proteasomes to leak into the extracellular space. As extracellular proteasomes have been shown to participate in a variety of cellular functions across cell types and disease states (Ben-Nissan et al., 2022), they could serve important roles in regulating protein clearance, especially of IDPs and damaged or oxidized proteins (Bonhoure et al., 2022), or in reducing neuroinflammation (Dianzani et al., 2019) by degrading pro-inflammatory cytokines in the extracellular milieu in the nervous system. Indeed, Dianzani et al. demonstrated that extracellular proteasomes can generate functional peptides and play a role in regulating cell migration and inflammation through cleavage of extracellular osteopontin, a cytokine implicated in diseases like multiple sclerosis (Dianzani et al., 2017; Dianzani et al., 2019).
Notably, early data has suggested differential regulation and expression of extracellular proteasomes and proteasome regulators found in EVs in neurodegenerative diseases including PD, raising the possibility of using proteasomal changes in EVs as a biomarker (Wang et al., 2019; Thompson et al., 2020). These results are encouraging for development of screening tools for neurogenerative disease and require more follow-up study. Critical questions about their functions remain, from regulation of their release to their role in healthy versus diseased brains (Dwivedi et al., 2021). While still in early stages, study of extracellular proteasomes in neurodegeneration has significant promise for understanding disease etiology and identifying novel therapies, including possibly for degradation of extracellular aggregates like amyloid-β plaques. The development of standardized, reliable tools for measuring their activity in the brain or spinal cord are needed. This recent review by Ben-Nissan et al. (2022) discusses the current understanding of the extracellular proteasome in depth, as well as experimental strategies to study this increasingly-recognized molecule.
Aging, oxidative stress, and protein aggregation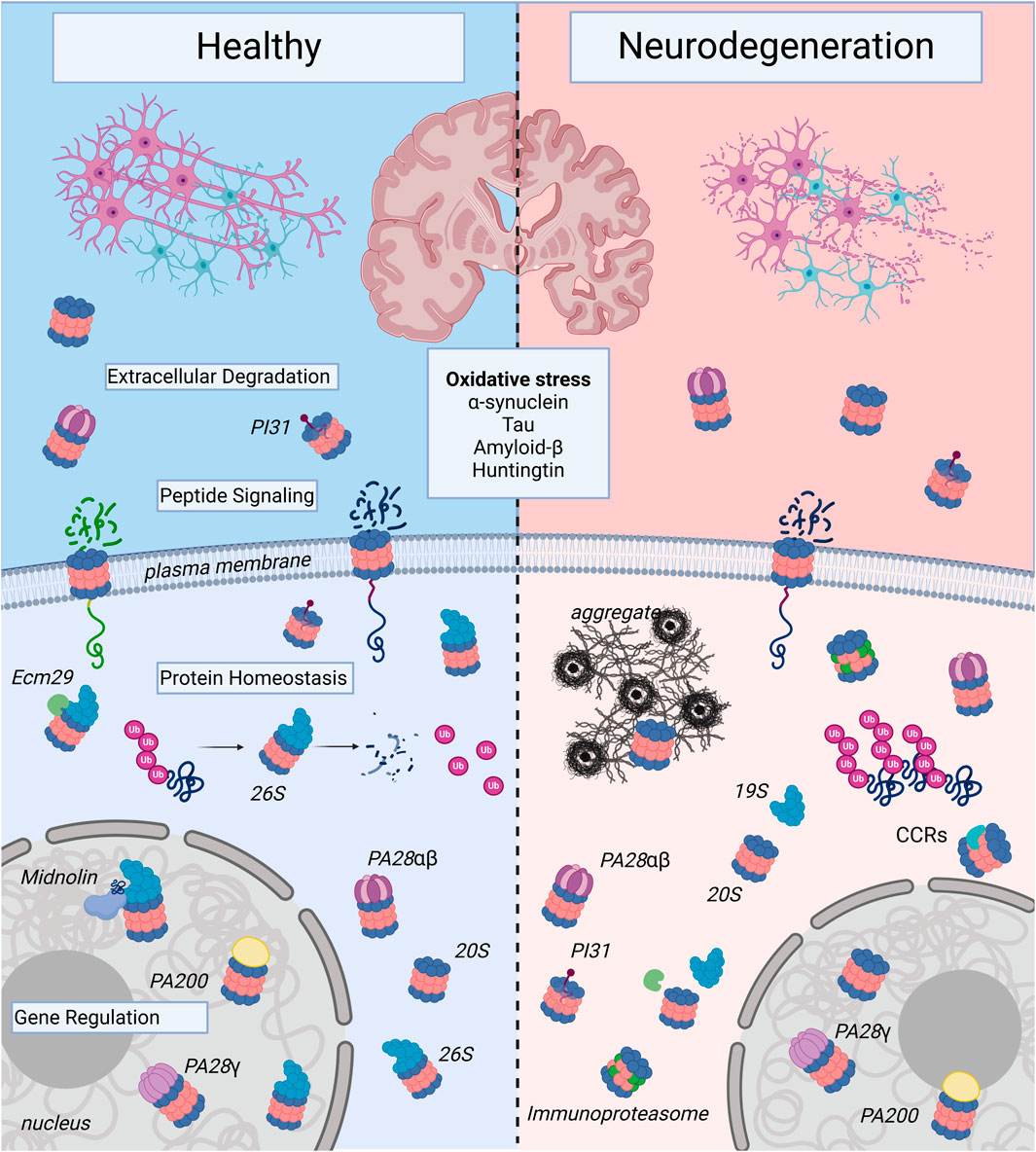
Figure 2. Proteasome Complexes in Health and Neurodegenerative Disease: The illustration shows healthy neurons and neurons affected by neurodegenerative disease. Note, the neurons undergoing neurodegeneration show fragmentation to illustrate decreased survival, which contributes to the decline in memory, cognition, and motor control seen in age-associated neurodegenerative illnesses such as Alzheimer’s Disease, Huntington’s Disease, and Parkinson’s Disease. At the molecular level, this inevitable cell death occurs in part due to proteasome dysregulation and is triggered by oxidative stress and toxic forms of aggregate-forming proteins such as α-synuclein, tau, amyloid-β, and huntingtin. In healthy neurons, there are a variety of proteasome complexes that exist under physiological conditions. In the nucleus, proteasomes with different proteasome activators (PAs; PA200, PA28γ, etc.) or uncapped 20S core particles (20S) have been demonstrated to regulate gene transcription. Cytosolic 20S proteasomes can also be activator-associated (e.g., 26S, PA28αβ) or uncapped. 26S proteasomes can mediate degradation of ubiquitinated proteins and produce small peptide fragments and free ubiquitin (Ub). In the extracellular space, there can be free as well as PA-capped 20S proteasomes, and in neurons, the 20S can be localized to the plasma membrane, where it serves a signaling function. In neurodegenerative diseases, proteasomes shift from predominantly ubiquitin-dependent degradation through the 26S to ubiquitin-independent degradation through alternatively-capped or uncapped 20S complexes. In addition, expression of immunoproteasome subunits is induced, particularly in the setting of chronic neuroinflammation. Proteasome complexes are regulated by adaptors like Ecm29, which mediates assembly and disassembly of the 26S proteasome from the 19S and 20S); PI31, a regulatory molecule that modulates proteasome activity from inside the 20S core; and catalytic core regulators (CCRs), which allosterically regulate the 20S to protect vital intrinsically-disordered proteins from degradation and serve critical roles in the oxidative stress response. Proteasomes are complex and heterogeneous molecules, and targeting different forms of the proteasome may prove useful for the development of preventative and disease-modifying therapies in neurodegeneration, an active area of research. Created in BioRender. Church, T. (2025) https://BioRender.com/k79b247.
AgingAs neurons age, damage to the proteome increases and overall neuronal proteasome activity decreases (Davidson and Pickering, 2023). Both the UPS and ubiquitin-independent proteasome function decline, further contributing to the aggregation of neurodegeneration-associated proteins such as tau, amyloid-β (Aβ), and α-syn (Bulteau et al., 2000; Keller et al., 2000b; Ding et al., 2006; Kelmer Sacramento et al., 2020; Cuanalo-Contreras et al., 2023; Davidson and Pickering, 2023). In addition, while glia normally secrete chaperones that assist neurons in maintaining proper protein folding, age-related 20S dysfunction could disrupt this process and further contribute to a decline in neuronal proteostasis (Chaplot et al., 2020; Leng and Edison, 2021).
With age, there is a shift from 26S to 20S proteasome activity concurrent with the decline in proteasome function, causing a relative increase in 20S activity (Keller et al., 2000b; Ding et al., 2006; Tonoki et al., 2009; Choi et al., 2023; Davidson and Pickering, 2023; Türker et al., 2023). Factors that contribute to this change include: oxidative stress, calpain activation, impaired assembly and recycling, and increased demand for IDP degradation (Huang et al., 2013; Coskuner-Weber et al., 2022; Davidson and Pickering, 2023). As aging is associated with increased oxidative stress and a decline in mitochondrial function, oxidation and damage to proteasome subunits can occur, especially subunits of the 19S cap (Reinheckel et al., 1998; Huang et al., 2013). Because 26S activity, assembly, and recycling are ATP-dependent, dysfunction of mitochondria also limits energy availability, further impairing 26S function and turnover. Furthermore, as oxidative damage accumulates, NADH, which stabilizes the 26S, oxidizes to its NAD+ form, compounding 19S dissociation from the 20S (Tsvetkov et al., 2014). As 26S activity declines, the 20S, which is more resilient to oxidative damage (Reinheckel et al., 1998), becomes more active in neurons by comparison, and its relative levels increase as association with the 19S cap decreases (Huang et al., 2013). Calpains, which are calcium-dependent proteases activated in aging and neurodegenerative diseases, cleave and inactive Rpn10, a 19S subunit, further decreasing 26S activity (Huang et al., 2013). Because aggregation-prone IDPs and damaged proteins with intrinsically disordered regions (IDRs) are preferentially degraded by the 20S rather than 26S, this shift to 20S activity may help to counteract the buildup of toxic protein aggregates over time in aging neurons (Opoku-Nsiah and Gestwicki, 2018; Davidson and Pickering, 2023).
Oxidative stressIn aging and neurodegenerative disease, a snowball effect of increasing oxidative stress, production of damaged and oxidized proteins, and inhibited proteasome activity can contribute to a progressively worsening cycle leading to neuronal dysfunction and cytotoxicity (Davies, 2001; Raynes et al., 2016). Sulfhydryl groups of the 19S PA are especially vulnerable to oxidation, causing 19S caps to lose their capacity to facilitate proteolysis and r
留言 (0)