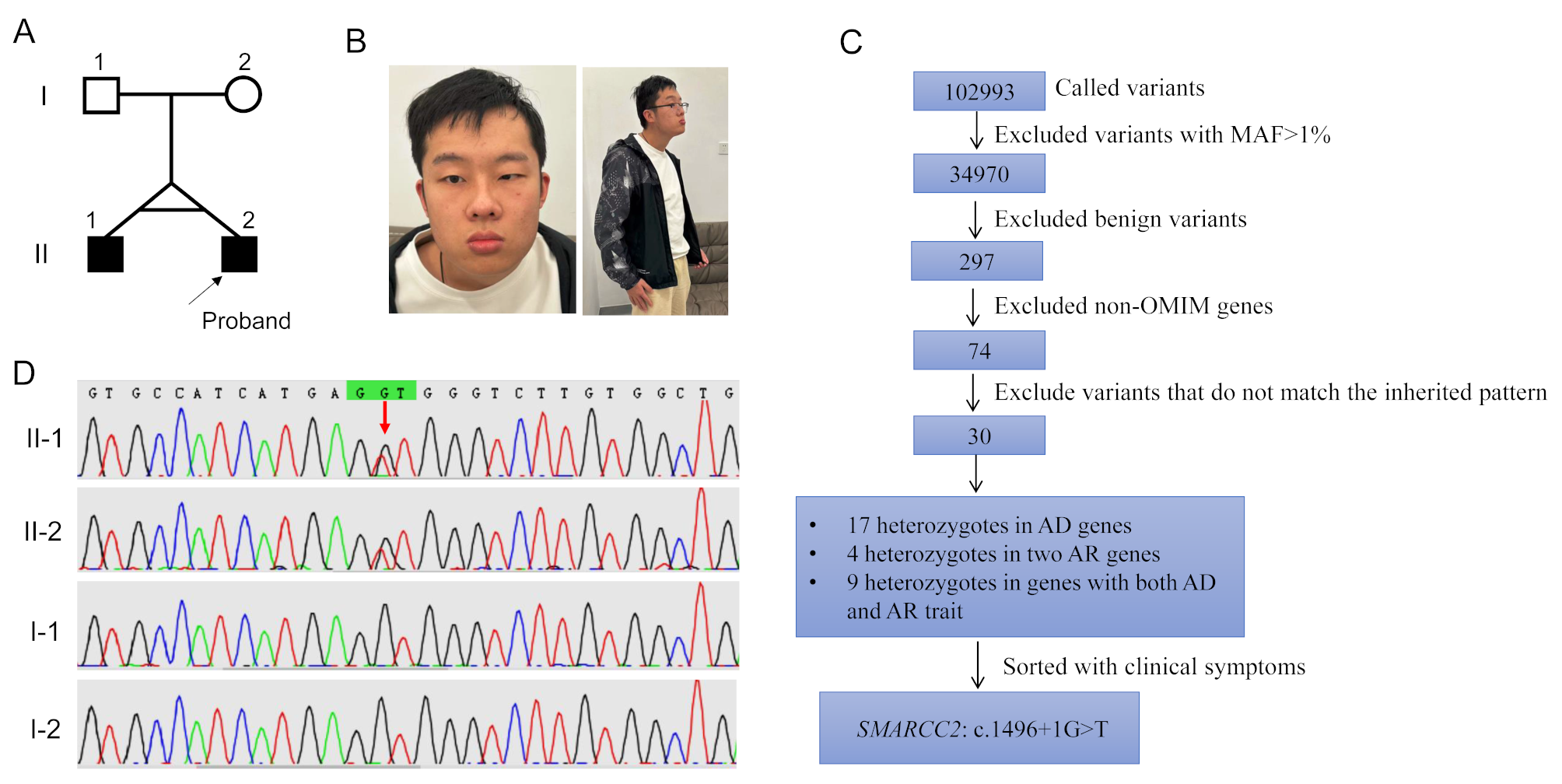
Herein, we described a 24-year-old proband with moderate DD/ID, language delay, cryptorchidism, hypotonia, behavioral abnormalities, sexual dysfunction, dense hair, craniofacial anomalies, allergic purpura and eczema, and drug allergies caused by a novel de novo splicing variant, c.1496 + 1G > T, in the SMARCC2 gene. His twin brother had the same genotype and most of the phenotypes were consistent. Transcriptional level analysis based on patient-derived samples showed that this canonical splicing variant causes two forms of splicing abnormalities: in-frame insertion of partial intron 16 or in-frame deletion of exon 16, which may escape NMD; this has already been observed in another splice donor variant, c.1833 + 1G > T [6]. Further structural analysis showed that in-frame changes caused by the c.1496 + 1G > T variant affected the assembly of the α-helices of the SMARCC2 protein. To the best of our knowledge, this is the first SMARCC2 mutation patient to seek medical assistance owing to reproductive needs. After a joint evaluation by a Reproductive Medicine and Genetics specialist, the proband was recommended third-generation in vitro fertilization (IVF) treatment.
The SMARCC2 protein contains five well-defined structural domains that are sequentially arranged from the N- to C-terminus as MarR-like (10–136), BRCT (140–183), Chromo (189–217), SWIRM (424–521), and SANT (596–647) domains (https://www.uniprot.org/uniprotkb/Q8TAQ2). The SWIRM domain mainly mediates specific protein-protein interactions [1, 24, 25], whereas the SANT domain is a chromatin-binding region of the protein that is thought to interact with unmodified histone tails [26]. In addition to the variants presented in this study, 45 NDD-related SMARCC2 variants have been reported to date, including 26 missense/in-frame and 29 truncating variants (e.g., nonsense/frameshift variants and splicing variants that result in out-of-frame changes of the protein). In addition to the variants reported in this study, four variants (c.1311-1G > A, c.1311–3 C > G, c.1496 + 1G > T, and p.Arg443Trp) in the SWIRM domain have been documented, and only c.1311–3 C > G was confirmed to cause NMD. Interestingly, missense and in-frame variants frequently occur in the SANT domain [16]. Notably, there were distinct phenotypic differences between the two patient groups. Among the patients with truncating variants, approximately 70% developed DD/ID, with the majority (76%) presenting with mild cases. In contrast, the missense/in-frame variant group had 100% incidence of DD/ID, with most cases being moderate-to-severe (81%). Additionally, patients with missense/in-frame variants had a higher incidence of hypotonia [16]. These results suggested that the SMARCC2 variant may not contribute solely to the disease via haploinsufficiency. For missense/in-frame variants, there may be additional pathogenic mechanisms, such as dominant-negative effects [16]. Although c.1496 + 1G > T is located in the SWIRM domain, our patient had moderate DD/ID and hypotonia, which further supports the conclusion that missense/in-frame variants of SMARCC2 are associated with a more severe neurological phenotype.
Bosch et al. summarized the clinical characteristics of 65 patients and found that it may be inappropriate to classify SMARCC2-related disease as CSS, particularly due to the absence of some representative features, such as hypoplasia of the finger/toenails or the absence of the distal phalanx of the fifth digit and prominent interphalangeal joints. These features were not observed in twin patients. Furthermore, the facial malformations in patients with SMARCC2 variants lack specificity. Among the 11 reported features, abnormalities in the external ear and thin upper lip vermilion occurred with a frequency exceeding 40% [16]. In our patients, in addition to the previously reported facial features of a prominent forehead, wide nose, low-set ears, and thin upper lip vermilion, we also discovered features of the epicanthus, which have rarely been described previously (Table 1). Therefore, we agree and recommend naming SMARCC2-related diseases separately and distinguishing them from CSS.
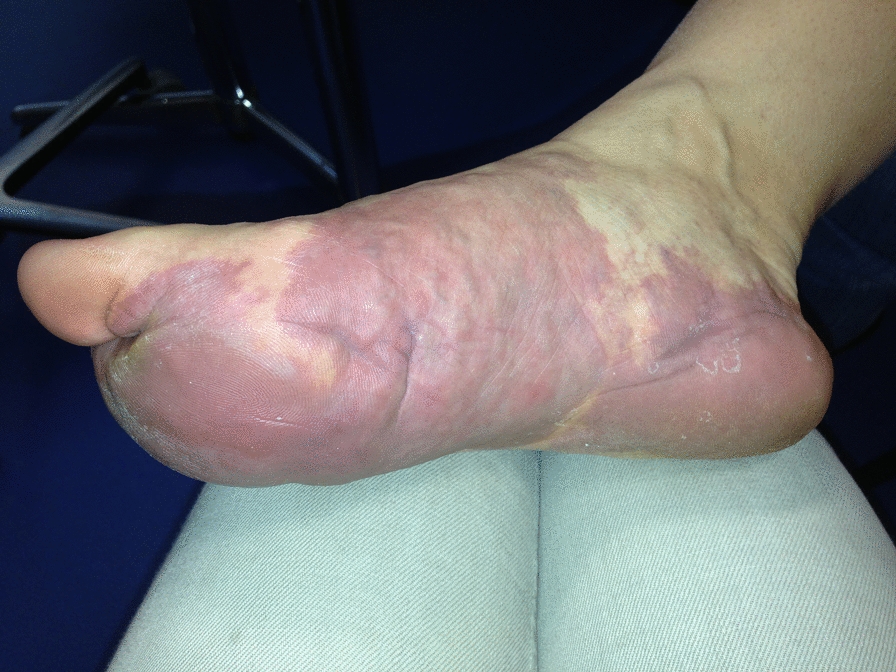
The phenotypes of most of our patients were consistent with those of previously reported patients, including core neurological features. However, we still found some previously unreported features, such as sexual dysfunction, allergic purpura, and drug allergies (Table 1). We particularly noticed the clinical features of erectile dysfunction in these twin patients because it may be an easily overlooked phenotype. In contrast, 12 of the 65 patients with SMARCC2 variants were adults, indicating that their assessment should be combined with developmental characteristics. This is particularly important because for most rare genetic diseases, we may not have the opportunity to collect data from adult patients; therefore, we generally focus on describing the medical history of pediatric patients. This may have caused us to miss the optimal assessment time, resulting in new risks for adult patients. A recent study of 35 adult patients showed that being overweight and obese were common in adult patients with CSS. In addition, the incidence of visual impairment, scoliosis, and behavioral abnormalities is significantly higher in adults [27]. Therefore, it is necessary to continuously follow-up the SMARCC2 mutant cohort, describe the clinical characteristics of all age groups, and draw a natural history of the disease.
In summary, we herein described a pair of twin adult patients harboring a novel splicing variant in the SWIRM domain of SMARCC2, which resulted in in-frame changes of the protein. Our findings reinforce the link between non-null variants of SMARCC2 and more severe neurological phenotypes, and underscore the need for distinct disease nomenclature in such cases. This study also highlights the importance of breaking away from the phenotypic framework of pediatric patients to conduct a comprehensive phenotypic assessment of adult patients harboring SMARCC2 variants.




留言 (0)