Subjects
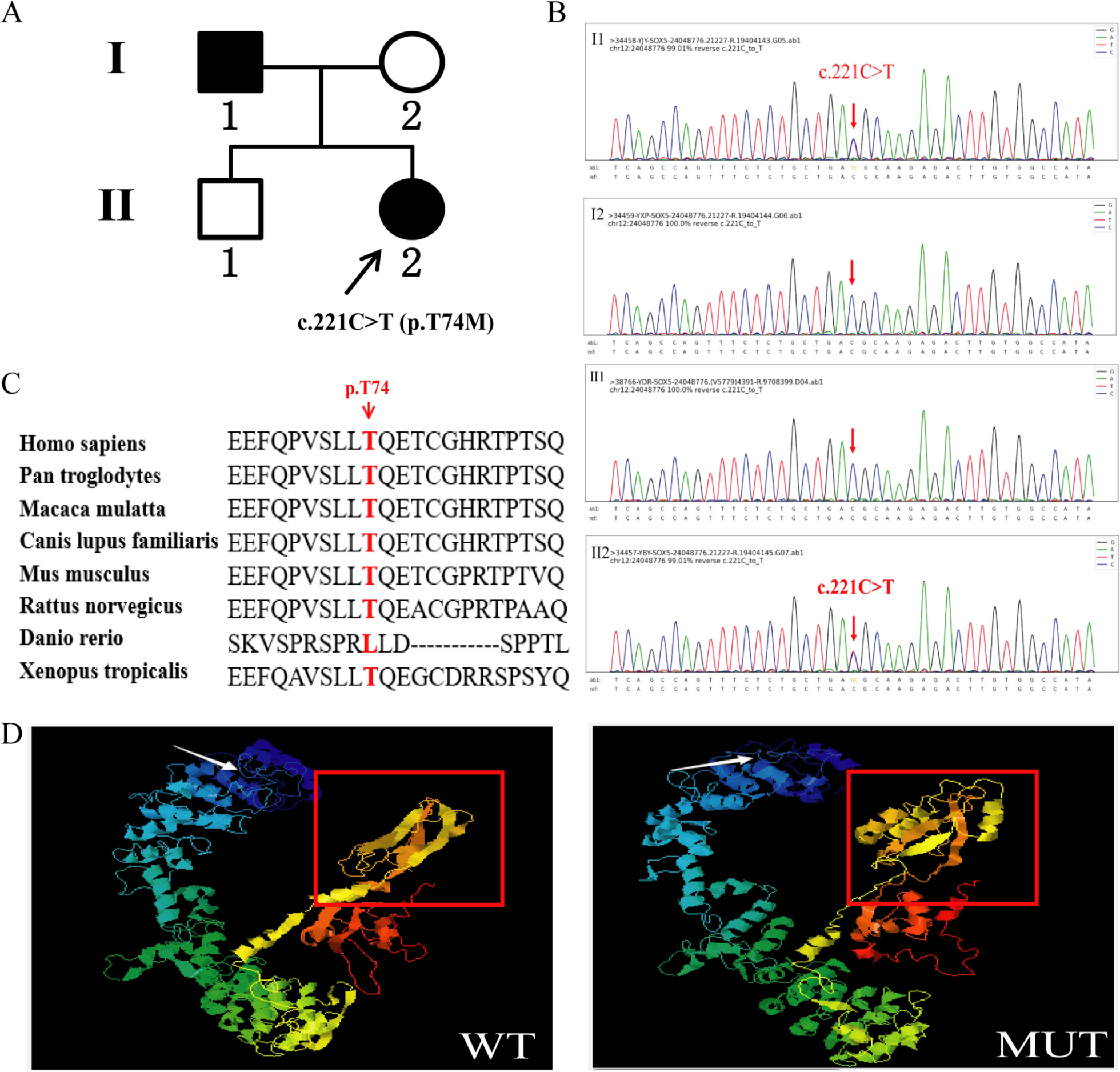
A normal adult male (II:1) presented to our hospital seeking genetic counseling, prompted by the presence of similar diseases in two family members (I:1 and II:2). Peripheral blood samples were collected from all participants, including two affected individuals (I:1 and II:2) and two unaffected individuals (I:2 and II:1). Comprehensive clinical examinations and high precision clinical exon sequencing were conducted at the Foshan Women and Children Hospital. The genetic analysis identified a variant in the SOX5 gene.
Variant analysis
Genomic DNA was extracted according to the standard operating instructions of the nucleic acid extraction or purification kit (AmCare Genomics Lab, Guangzhou, China). Then the DNA was digested and fragments were modified by adding an “A” base at the 3 ‘end and ligated to the adaptors. Custom-designed Amcare probes (AmCare Genomics Lab, Guangzhou, China) were used for in-solution hybridization to enrich target sequences, which included coding exons for about 5000 clinically relevant disease-causing genes. The genes were selected based on reports in OMIM, HGMD, and peer-reviewed literature. Known pathogenic variants in deep intronic and other non-coding regions in targeted genes were also included. The libraries of genomic DNA samples were prepared using the Gene Sequencing Library Kit (Random Endonuclease Method) (AmCare Genomics Lab, Guangzhou, China), then adaptors were added and they were amplified with pre-capture ligation-mediated PCR (LM-PCR). The quality and fragment size of the DNA samples in the library were assessed using 1.0% agarose gel electrophoresis. The multifunctional microplates (Molecular Devices) were used to detect the concentration of the library. Enriched DNA samples were indexed and sequenced on the AmCareSeq-2000 sequencer (AmCare Genomics Lab, Guangzhou, China). The average coverage depth was 200 × with > 98% of the target regions covered by at least 20 reads.
Bioinformatics
The raw reads in fastq format were filtered for adapter sequences, polyN, polyA, and other sequences using the fastp software, and low-quality reads (QC less than 20) were removed to obtain clean reads for subsequent data analysis. The sequencing reads were then mapped to the reference human genome version hg19 using Burrows-Wheeler Aligner (BWA, v0.7.15). Harmful and pathogenic variants were predicted by Mutation Taster (https://www.mutationtaster.org/), PROVEAN and SIFT (http://provean.jcvi.org/protein_batch_submit.php?species=human). The protein stability was predicted by I-Mutant (https://folding.biofold.org/i-mutant/i-mutant2.0.html). The three-dimensional (3D) protein structures of wild-type and mutant SOX5 proteins were predicted using I-TASSER (https://zhanggroup.org//I-TASSER/). The sequences of several species were compared to the reference sequence from the NCBI database (https://www.ncbi.nlm.nih.gov/homologene/) to understand the conservation of the variant site across different species.
SOX5 expression plasmid constructs and mutagenesis
Total RNA was isolated from K562 cells using RNAex Pro Reagent (Accurate Biology, Hunan, China). The first-strand cDNA was then synthesized using the HiScript II 1st Strand cDNA Synthesis Kit (Vazyme, Jiangsu, China). The SOX5 coding sequence was amplified using the Table 1 primers, designed with EcoRI and BamHI restriction sites. After double digestion with EcoRI and BamHI (New England Biolabs, Beijing, China), the PCR products were cloned into the pEGFP-N1 vector. The c.211C > T variant was amplified by PCR site-directed mutagenesis, using wild-type plasmid as template. The site-directed mutagenic primers were designed as shown in Table 1. Recombinant plasmids were purified using the EndoFree Mini Plasmid Kit II (TIANGEN, Beijing, China), followed by Sanger sequencing to identify the wild-type and mutant.
Table 1 Sequences of the primers used for PCRRNA analysis
Human embryonic kidney (HEK) 293T cells and Neuro-2a cells, at 70% confluence, were transfected with 1 μg of the recombinant plasmids containing wild-type (pEGFP-N1-SOX5-WT) or mutant SOX5 c.221C > T genes (pEGFP-N1-SOX5-MUT), using Lipofectamine™ 3000 transfection reagent (Invitrogen, NY, USA). After 24 h of transfection, total RNA was isolated using the RNAex Pro Reagent (Accurate Biology, Hunan, China) and reverse transcribed into cDNA using the HiScript III RT SuperMix for Quantitative Real-time PCR (qPCR)(+ gDNA wiper) (Vazyme, Nanjing, China). The relative mRNA levels of wild-type and mutant SOX5 were then measured using the ChamQ SYBR qPCR Master Mix (Vazyme, Nanjing, China). We overexpressed wild-type and mutant SOX5 and then tested the expression levels of the ACAN, AXIN2, SOX9 and PDGFRA genes. The GAPDH gene was used as the reference gene to normalize expression. These qPCR primers were shown in Table 1. The 2-ΔΔCt method was used to calculate the levels of gene expression. Transfection and real-time PCR assays were repeated three times to confirm the reproducibility of the results.
Western blotting analysis and protein stability
To analyze the difference in protein levels between the wild-type and mutant SOX5 cells, Neuro-2a cells and 293T cells were transfected with 2.5 μg of pEGFP-N1, pEGFP-N1-SOX5-WT or pEGFP-N1-SOX5-MUT vector using Lipofectamine™ 3000 transfection reagent. Forty-eight hours after transfection, cells were collected, washed with cold PBS (Sigma-Aldrich) and lysed with cell lysis buffer (Beyotime Biotechnology, Shanghai, China) that contained 1% phenylmethanesulfonyl fluoride (Beyotime Biotechnology). After shaking for 30 min at 4 °C, cell debris was removed by centrifugation. We added 5 × sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (Beyotime Biotechnology, Shanghai, China) to the cell lysates before boiling for 5–10 min. Protein samples were then separated by a 10% SDS–polyacrylamide gel and transferred onto a polyvinylidene fluoride membrane (Millipore, MA, USA). After being blocked for one hour with 5% skimmed milk at room temperature, the membranes were incubated overnight at 4 °C with green fluorescent protein (GFP)-tagged mouse monoclonal antibodies (proteintech, Rosemont, USA), Cyclin A polyclonal antibodies (Bioworld, Minnesota, USA) or HRP GAPDH mouse monoclonal antibodies (proteintech, Rosemont, USA). The following day, membranes were incubated with goat anti-mouse IgG or goat anti-Rabbit IgG (proteintech, Rosemont, USA) at room temperature for 1.5 h after three washes with Tris-Buffered Saline-Tween 20. The immunoreactive proteins were visualized with SuperSignal West Pico ECL (Thermo Fisher Scientific) and a digital chemiluminescence system (Tanon Science & Technology, Shanghai, China). Additionally, to study SOX5 protein degradation, at 48 h post-transfection, 100 µg/ml of cycloheximide (Sigma-Aldrich) was added to each well. Total cell protein was collected at 0, 2, 4, 6, and 8 h to evaluate the half-life of the SOX5 protein. Western blot was used to analyze and evaluate the half-life of SOX5 expression. The protein levels were quantified using ImageJ software. Protein analysis was repeated three times for each test and SOX5 and Cyclin A levels were normalized to GAPDH.
Subcellular localization
The HEK 293T cells and Neuro-2a cells were cultured in DMEM (Gibco, NY, USA) with 10% fetal bovine serum (Gibco, NY, USA) at 37 °C and under 5% CO2. The pEGFP-N1-SOX5-WT and pEGFP-N1-SOX5-MUT vectors were transfected into the HEK 293T and Neuro-2a cells using Lipofectamine 3000 transfection reagent. After transfection for 36 h, cells were washed three times with PBS and fixed for 30 min with 4% paraformaldehyde at room temperature. After fixation, cells were washed three times with PBS and incubated in 0.2% Triton X-100 (Thermo Fisher Scientific, MA, USA) to increase the permeability of the cytomembrane. Finally, the nuclei were stained with DAPI (Beyotime Biotechnology, Shanghai, China). An inverted confocal fluorescence microscope (LSM 880; Carl Zeiss AG, Jena, Germany) was used for imaging the cells.
Cell cycle flow cytometry and CCK-8 cell proliferation assay
The pEGFP-N1-SOX5-WT and pEGFP-N1-SOX5-MUT vectors were transfected into Neuro-2a cells using Lipofectamine 3000 transfection reagent. After transfection for 36 h, the cells were collected using trypsin digestion and perfect medium termination. Cells were washed with PBS and fixed overnight with 70% ethanol at 4 °C and then washed again with PBS. They were then incubated with propidium iodide staining solution (Beyotime Biotechnology, Shanghai, China) at 37 °C in a dark environment for 30 min. After staining, cells were stored at 4 °C or in an ice bath away from light. Red fluorescence and light scattering were detected by flow cytometry at an excitation wavelength of 488 nm. The Flowjo software was used for cell DNA content analysis and light scattering analysis.
The CCK-8 assay kit was used to assess cell proliferation. Neuro-2a cells were first transfected with the pEGFP-N1-SOX5-WT and pEGFP-N1-SOX5-MUT vectors using Lipofectamine 3000 transfection reagent. The transfection system was set up in a 12-well plate, divided into three groups: the first group was transfected with 1 μg of WT per well, the second group with 1 μg of MUT per well, and the third group was co-transfected with 500 ηg each of WT and MUT per well, for a total of 1 μg. After 24 h of transfection, 10^4 cells per well were seeded evenly into a 96-well plate and cultured for 12, 24, and 36 h. Subsequently, 100 μl of complete medium containing 10 μl of CCK-8 solution was added to each well, followed by incubation at 37 °C for 1.5 h. Absorbance at 450 nm was then measured using a microplate reader.
Statistical analyses
GraphPad Prism software was used for statistical analyses. The independent samples T test and two-way ANOVA were used to determine the statistical significance between two groups. Statistical data were expressed as the mean ± standard deviation (SD). A P value < 0.05 was considered to be significant. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.




留言 (0)