
記住我
NO-containing compounds have been used in medicine for over 160 years; however, it was not until the early 1980s that NO was discovered as the ingredient that exerts the therapeutic effects and ECs as the key source of vascular NO (Katsuki et al., 1977; Arnold et al., 1977; Furchgott and Zawadzki, 1980; Ignarro et al., 1987; Palmer et al., 1987). We now know NO can be produced by three NO synthases and via nitrate-nitrite-NO conversion (Bredt et al., 1991; Janssens et al., 1992; Geller et al., 1993; Benjamin et al., 1994; Lundberg et al., 1994; Stuehr, 1997). The NO synthases are classified a neuronal NOS (encoded by NOS1), inducible NOS (encoded by NOS2), and eNOS (encoded by NOS3). Among these sources, endothelium-derived NO plays a vital role in regulating vascular tone, inhibiting inflammation, and preventing thrombosis (Alheid et al., 1987; Forstermann et al., 1989; Bredt and Snyder, 1990; Mitchell et al., 1991; Forstermann et al., 1991). Endothelium-derived NO diffuses into the circulation and the underlying VSMCs, where it activates sGC, which enhances cGMP, causing vasodilation (Arnold et al., 1977; Ignarro et al., 1986a; Ignarro et al., 1986b; Forstermann et al., 1994). The critical role of eNOS in controlling vascular tone was documented by findings that pharmacological inhibition of NOS causes hypertension (Rees et al., 1989) and deletion of NOS3 results in high blood pressure (Huang et al., 1995). Beyond activating sGC and enhancing cGMP, NO exerts numerous other effects. For example, endothelium-derived NO directly suppresses the electrical excitability of VSMCs by inhibiting the action of T-type and L-type voltage-gated Ca2+ channels in small arteries, and reduced NO availability can trigger transient depolarization in normally quiescent VSMCs leading to vasospasm (Smith et al., 2020). Actions of NO leading to smooth muscle relaxation are important for cardiovascular, respiratory, renal and digestive functions. NO is also critical in brain function as a neurotransmitter and immune responses.
Efforts to increase NO bioavailabilityDysfunction and uncoupling of eNOS are associated with cardiovascular diseases (CVD) (Janaszak-Jasiecka et al., 2023; Heitzer et al., 2000a; Munzel et al., 2005), and increased eNOS expression and reversal of eNOS uncoupling in experimental models improves vascular function (Li et al., 2006). Approaches to increase NO bioavailability have been intensively researched. Some studies suggest that diets that are high in antioxidants or antioxidant supplementation can help preserve vascular health and prevent CVD by reducing oxidative stress and improving endothelial function. However, a blanket recommendation has not been made in clinical guidelines as there are needs for stronger evidence and determinations of effective doses and specific patient populations that might benefit (Varadharaj et al., 2017). Dietary nitrates and nitrites, found in foods such as beetroot and leafy greens, can be converted to NO and have shown promising results in improving vascular function and lowering blood pressure. However, these are not yet recommended in clinical guidelines for prevention or management of CVD (Blekkenhorst et al., 2018). Current options in clinical practice guidelines focus mainly on downstream components of NO signaling, such as NO inhalation, use of NO donors, sGC stimulators/activators, or inhibition of phosphodiesterase (PDE) 5 (Chen et al., 2002; Rapoport et al., 1987; Bonderman et al., 2014; Ignarro et al., 1982; Kraehling and Sessa, 2017; Lundberg et al., 2015). However, challenges such as short-lived effects and the development of nitrate tolerance and endothelial dysfunction have limited their efficacy (Rapoport et al., 1987; Munzel et al., 1995a; Munzel et al., 1995b; Erdmann et al., 2013; Knorr et al., 2011; Munzel et al., 2014; Munzel et al., 2013; Oelze et al., 2013). There is thus a strong need to develop new and improve existing approaches to promote the endogenous production of NO.
The regulation of eNOSeNOS is a ∼133 kDa homo-dimeric oxidoreductase enzyme with an N-terminal oxygenase domain that binds L-arginine and tetrahydrobiopterin (BH4) and a C-terminal reductase domain that transfers electrons from nicotinamide adenine dinucleotide phosphate (NADPH) via flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) (Figure 1) (Stuehr, 1997; Palmer et al., 1988; Stuehr et al., 2005; Marsden et al., 1992). Dimerization is essential for eNOS function, stabilizing the enzyme and ensuring efficient electron transfer (List et al., 1997). Ca2+-bound calmodulin (CaM) binds to the CaM-binding domain and initiates electron flow from the reductase domain to the oxygenase domain, where NO is synthesized from L-arginine (Busse and Mulsch, 1990; Venema et al., 1996). eNOS-mediated production of NO follows a two-step process: 1) hydroxylation of L-arginine to Nω-hydroxy-L-arginine, and 2) further oxidation to generate L-citrulline and NO. Tetrahydrobiopterin (BH4) plays a vital role in this reaction by stabilizing the eNOS dimer and preventing the formation of superoxide, a potentially harmful byproduct, ensuring NO synthesis proceeds efficiently (Xia et al., 1998a; Xia et al., 1998b). eNOS regulation is a highly complex process, integrating multiple layers of control to fine-tune NO production according to physiological needs. These layers include transcriptional regulation, post-translational modifications, and protein-protein interactions.
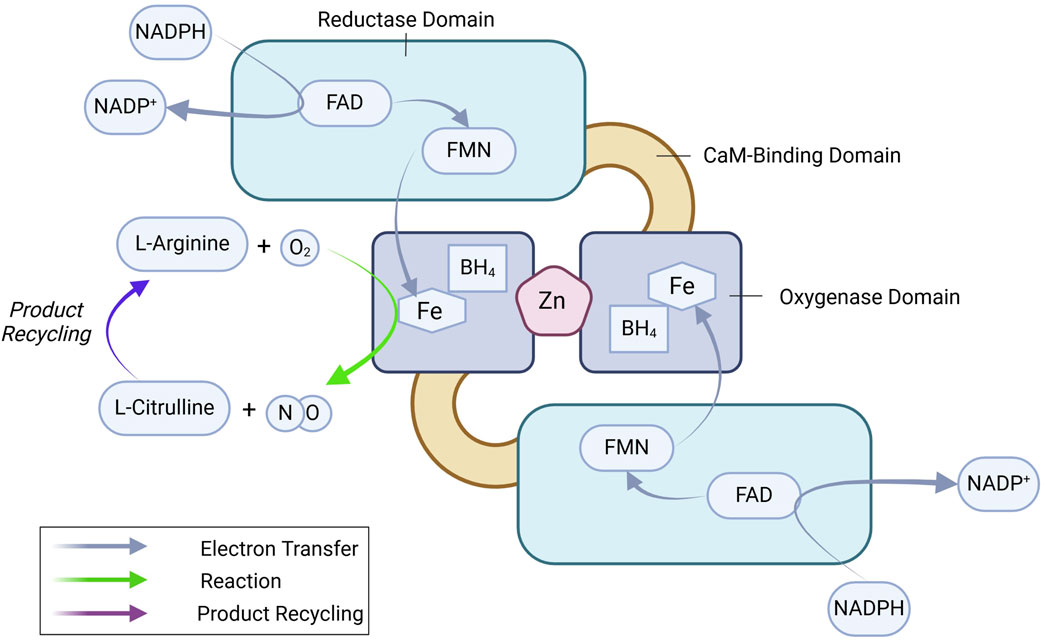
Figure 1. eNOS dimer and key enzymatic reactions leading to NO production. See text for details. BH4, tetrahydrobiopterin; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; NADPH, nicotinamide adenine dinucleotide phosphate.
Transcriptional regulation of eNOSThe NOS3 gene is located on chromosome 7 (7q35-36) and is regulated by a promoter region containing binding sites for multiple transcription factors, including Krüppel-like Factor 2 (KLF2), Specificity protein 1 (Sp1), Specificity protein 3 (Sp3), Ets1, mothers against decapentaplegic homolog-2 (Smad2), and Nuclear factor erythroid 2-related factor (Nrf2), among others (Karantzoulis-Fegaras et al., 1999; Laumonnier et al., 2000; Balligand et al., 2009; Fish and Marsden, 2006). These factors dynamically regulate eNOS expression in response to physiological signals. Initially thought to be constitutively expressed, NOS3 is now recognized to be highly responsive to various regulatory stimuli, which adjust its transcription to match the vascular environment’s needs (Balligand et al., 2009; Black et al., 1998; Mattsson et al., 1997; Nadaud et al., 1996; Nishida et al., 1992; Ou et al., 2005; Searles, 2006). Transcriptional regulation of eNOS can be categorized into upregulating, downregulating, and dual-regulating factors (Figure 2).
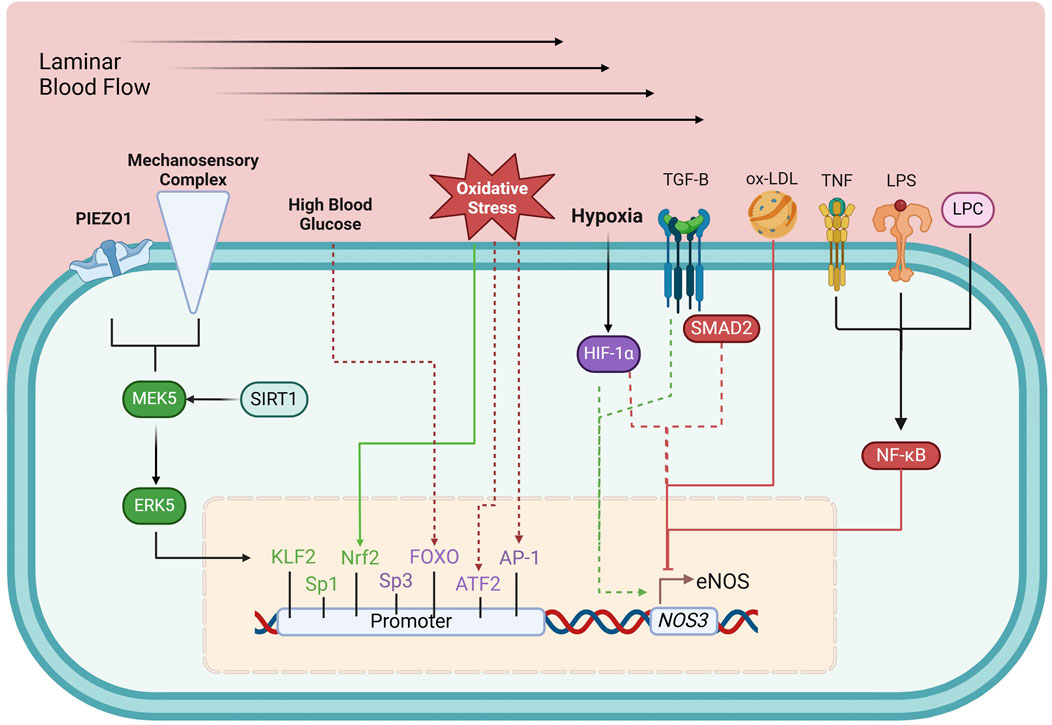
Figure 2. Transcriptional regulation of eNOS. Green boxes, upregulating factors; purple boxes, dual-regulating factors; red boxes, down-regulating factors; solid arrows, connection between environmental and metabolic elements to their respective regulating factors; dashed arrows, circumstantial elements that determine the effects of dual-regulating factors; AP-1, activator protein 1; ATF-2, activating transcription factor 2; ERK5, extracellular-regulated kinase 5; FOXO, forkhead box O; HIF-1α, hypoxia-inducible factor 1-α; KLF2, Krüppel-like factor 2; LPC, lysophosphatidylcholine; LPS, lipopolysaccharide; MEK5, Mitogen-activated protein kinase kinase 5; Nrf2, Nuclear factor erythroid 2-related factor; NF-κB, Nuclear Factor kappa-light-chain-enhancer of activated B cells; ox-LDL, oxidized low-density lipoprotein; SIRT1, sirtuin 1; SMAD2, mothers against decapentaplegic homolog-2; Sp1, specificity protein 1; Sp3, specificity protein 3; TGF-β, tumor growth factor β; TNF, tumor necrosis factor. See text for details.
Upregulating factorsShear stress, the mechanical stimulus exerted on the endothelium by laminar blood flow, upregulates eNOS expression (Nadaud et al., 1996) by triggering PIEZO1 Ca2+ channels (Wang et al., 2016) and a mechanosensory complex leading to the activation of KLF2, which binds directly to the NOS3 promoter (Wang et al., 2010; Lin et al., 2005). Sp1 regulates basal NOS3 expression and responds to stimuli like growth factors and hypoxia, enhancing NO production to maintain vascular tone (Tang et al., 1995; Wariishi et al., 1995). Additionally, Nrf2, activated by oxidative stress, enhances NOS3 transcription by upregulating antioxidant response elements in the NOS3 promoter (Wu et al., 2019).
Downregulating factorsNF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) represses eNOS expression, particularly in inflammation. NF-κB is activated by pro-inflammatory stimuli such as tumor necrosis factor-alpha (TNF-α), lipopolysaccharide (LPS), and oxidized low-density lipoprotein (ox-LDL). These molecules trigger signaling events that lead to NF-κB translocation into the nucleus, where it inhibits eNOS transcription (Nishida et al., 1992; Neumann et al., 2004). LPS and ox-LDL, both associated with oxidative stress and vascular inflammation, promote NF-κB activation, further repressing eNOS expression (Liao et al., 1995; Lu et al., 1996).
Dual-regulating factorsCertain factors can both up- or downregulate eNOS expression depending on cellular conditions. Tumor growth factor β (TGF-β)/Smad2 can enhance eNOS transcription in a healthy endothelium but suppress it in chronic inflammation or vascular injury (Saura et al., 2002). Hypoxia-inducible factor-1α (HIF-1α) also exhibits dual regulation: during acute hypoxia, it stimulates eNOS expression to ensure adequate NO production but may suppress eNOS in chronic hypoxia, causing maladaptive vascular changes (McQuillan et al., 1994; Fish et al., 2010).
Post-transcriptional mechanisms offer additional precision by modulating the stability and translation of eNOS mRNA. Elements such as miRNAs (e.g., miR-92a) and long non-coding RNAs (lncRNAs) can either enhance or suppress NO production in response to physiological conditions (Lin et al., 2005; Miao et al., 2018; Suarez et al., 2007; Man et al., 2018; Bonauer et al., 2009).
Post-translational modifications (PTMs) of eNOSeNOS activity is also intricately regulated by various PTMs, including phosphorylation, acetylation, S-nitrosylation, and palmitoylation. These modifications play critical roles in modulating eNOS enzymatic activity, localization, and interactions with other cellular components (Figure 3).
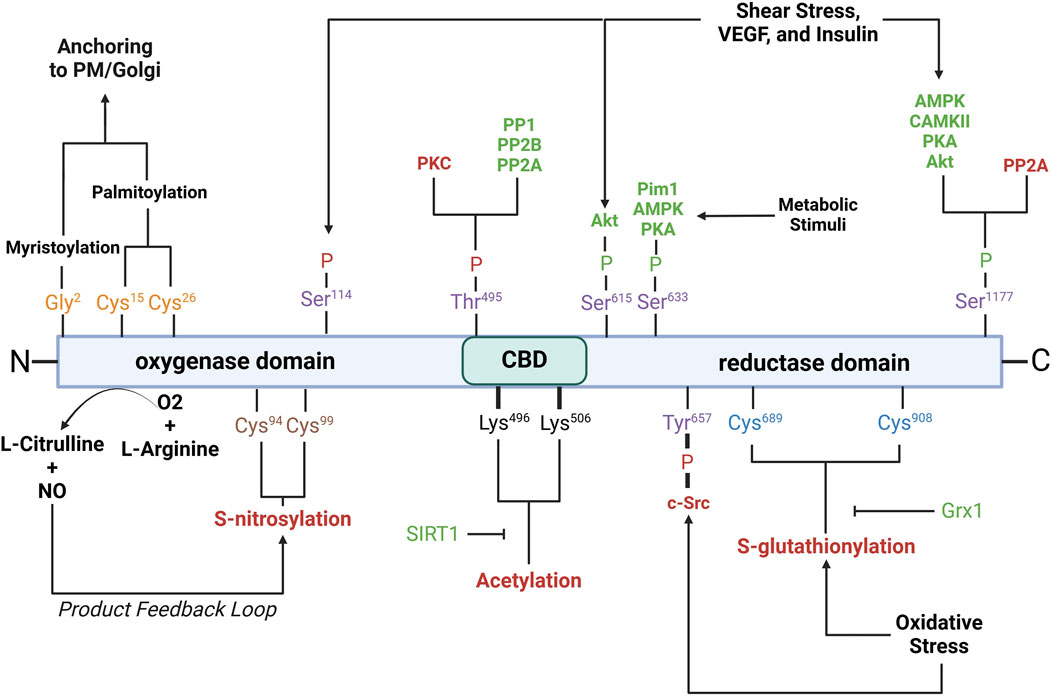
Figure 3. Schematic of eNOS domains with key residues involved in post-translational modifications. Purple residues, phosphorylation sites; yellow residues, myristoylation/palmitoylation sites; brown residues, S-nitrosylation sites; black residues, acetylation sites; blue residues, S-glutathionylation sites. Red- and green-letter enzymes, inhibitory and stimulatory effects on eNOS, respectively. Akt, protein kinase B; AMPK, AMP-activated protein kinase; CaMKII, calcium/calmodulin-dependent protein kinase II; c-Src, cellular sarcoma; Grx1, glutaredoxin; Pim1, proviral integration site for Moloney murine leukemia virus 1; PKA, protein kinase A; PKC, protein kinase C; PP1, protein phosphatase 1; PP2A, protein phosphatase 2a; PP2B, protein phosphatase 2b; PM, plasma membrane; SIRT1, sirtuin 1; VEGF, vascular endothelial growth factor. See text for further details.
Phosphorylation is a critical modification that modulates eNOS activity and is tightly regulated by various kinases and phosphatases in response to physiological cues such as shear stress, hypoxia, and growth factors. Serine 1177 is located on the C-terminal reductase domain and when phosphorylated, enhances eNOS activity by facilitating electron flow from NADPH to the heme domain, contributing to NO production (Fulton et al., 1999; Scotland et al., 2002; Kashiwagi et al., 2013; Li Q. et al., 2013; Park et al., 2016). Ser1177 phosphorylation is key positive regulator of eNOS function (Dimmeler et al., 1999; Tomada et al., 2014). Stimuli such as shear stress, vascular endothelial growth factor (VEGF), and insulin activate kinases including protein kinase B (Akt), AMP-activated protein kinase (AMPK), calcium/calmodulin-dependent protein kinase II (CaMKII), protein kinase A (PKA), and protein kinase G (PKG), which phosphorylate Ser1177 (Fulton et al., 1999; Dimmeler et al., 1999; Michell et al., 1999; Chen et al., 1999; Fleming et al., 2001; Atochin et al., 2007). Phosphorylation by Akt, in particular, is essential for eNOS activation in endothelial cells in response to VEGF and shear stress (Dimmeler et al., 1999; Di Lorenzo et al., 2013). Ser1177 phosphorylation also increases the Ca2+ sensitivity of the synthase, permitting CaM binding and enzyme activation at lower intracellular Ca2+ levels (Tran et al., 2009; Mount et al., 2007; McCabe et al., 2000). Serine 633 is phosphorylated in response to shear stress, exercise, and metabolic stimuli (Mount et al., 2007). Ser633 phosphorylation by PKA and AMPK during physical activity improves NO bioavailability and supports vascular homeostasis (Mount et al., 2007; Michell et al., 2002). Serine 615 phosphorylation enhances eNOS activity, working cooperatively with Ser1177 to enhance the binding affinity of the Ca2⁺-CaM complex to eNOS, a critical step for eNOS activation, which ensures a robust response to Ca2⁺ signals (Tran et al., 2009; Bauer et al., 2003). Threonine 495 phosphorylation, in contrast, inhibits eNOS by suppressing CaM binding (Fleming et al., 2001). Kinases like AMPK and PKC mediate this modification, especially during oxidative stress (Chen et al., 1999). Agonists such as bradykinin promote NO release by inducing Thr495 dephosphorylation, allowing CaM to activate eNOS (Harris et al., 2001). This dephosphorylation is mediated by calcineurin and inhibited by cyclosporine A (Harris et al., 2001). The balance between Thr495 phosphorylation and dephosphorylation is crucial for regulating eNOS activity and NO production. Serine 114 is phosphorylated in response to shear stress (Mount et al., 2007; Gallis et al., 1999). Though its role remains debatable, phosphorylation at Ser114 is considered a negative regulator of eNOS activity (Mount et al., 2007), supported by the observations that its dephosphorylation by VEGF treatment enhances eNOS function (Bauer et al., 2003) and that a phospho-null mutation here inhibits eNOS activity (Kou et al., 2002; Li et al., 2007).
Acetylation regulates eNOS interactions with other proteins, its plasma membrane localization, and overall enzymatic efficiency. Acetylation at lysine 609 affects eNOS interaction with heat shock protein 90 (Hsp90) and CaM, both essential for eNOS activation (Taubert et al., 2004). Lysine 609 acetylation is mediated by histone deacetylase 3 and inhibits eNOS activity by preventing proper electron transfer (Jung et al., 2010). In contrast, its deacetylation by sirtuin 1 (SIRT1) restores eNOS activity, enhancing eNOS-CaM interaction (Donato et al., 2011; Arunachalam et al., 2010).
S-nitrosylation is a reversible modification that constrains NO synthesis via a product feedback mechanism (Lipton et al., 1993; Erwin et al., 2005; Lima et al., 2010). S-nitrosylation involves the covalent attachment of a NO group to cysteine thiols, specifically Cys94 and Cys99 of eNOS, forming S-nitrosothiols (SNOs) (Erwin et al., 2006). Cys94 and Cys99 are located within the zinc tetrathiolate cluster (Erwin et al., 2005) that is important for the eNOS dimer interface; nevertheless, mutation of these sites does not disrupt dimer formation (Erwin et al., 2005). Paradoxically, agonist stimulation, which increases NO production, also promotes rapid denitrosylation of eNOS, in a similar timeframe as phosphorylation at Ser1177 (Erwin et al., 2005). S-nitrosylated eNOS exhibits reduced catalytic activity, which can be reversed with the release of NO. The subcellular localization of eNOS influences the degree of S-nitrosylation, with membrane-bound eNOS being more heavily nitrosylated than its cytosolic counterpart due to higher NO production at the membrane (Erwin et al., 2006).
Glutathionylation is a reversible post-translational modification in which the tripeptide glutathione attaches to cysteine residues in eNOS, notably Cys689 and Cys908, in the reductase domain (Chen et al., 2010). Glutathionylation is promoted by oxidative stress and results in decreased NO production and increased superoxide generation due to disrupted flavin-dependent electron transport (Chen et al., 2010; Crabtree et al., 2013). Fortunately, this modification is reversible through the action of glutaredoxin (Grx1), which interacts closely with eNOS (Chen et al., 2013). Loss of Grx1, either by oxidative stress or genetic silencing, increases eNOS glutathionylation and further NO synthesis (Chen et al., 2013).
Palmitoylation and myristoylation are lipid modifications that regulate the localization and activity of eNOS. Myristoylation, the irreversible attachment of myristic acid to Gly2, anchors eNOS to membranes such as the plasma membrane and Golgi apparatus (Liu et al., 1995; Sessa et al., 1995). Myristoylation is a prerequisite for palmitoylation, a reversible process where palmitic acid binds to Cys15 and Cys26, anchoring eNOS within plasmalemmal caveolae (Fernandez-Hernando et al., 2006). The reversible cycle of palmitoylation and depalmitoylation allows eNOS to dynamically shift between membrane locations in response to physiological signals (Yeh et al., 1999).
Regulation of eNOS by protein-protein interactions (PPIs)eNOS activity is intricately regulated through its interactions with various binding partners. These interactions play essential roles in modulating its localization, dimerization, and activation, ensuring that eNOS responds appropriately to cellular and environmental signals (Figure 4).
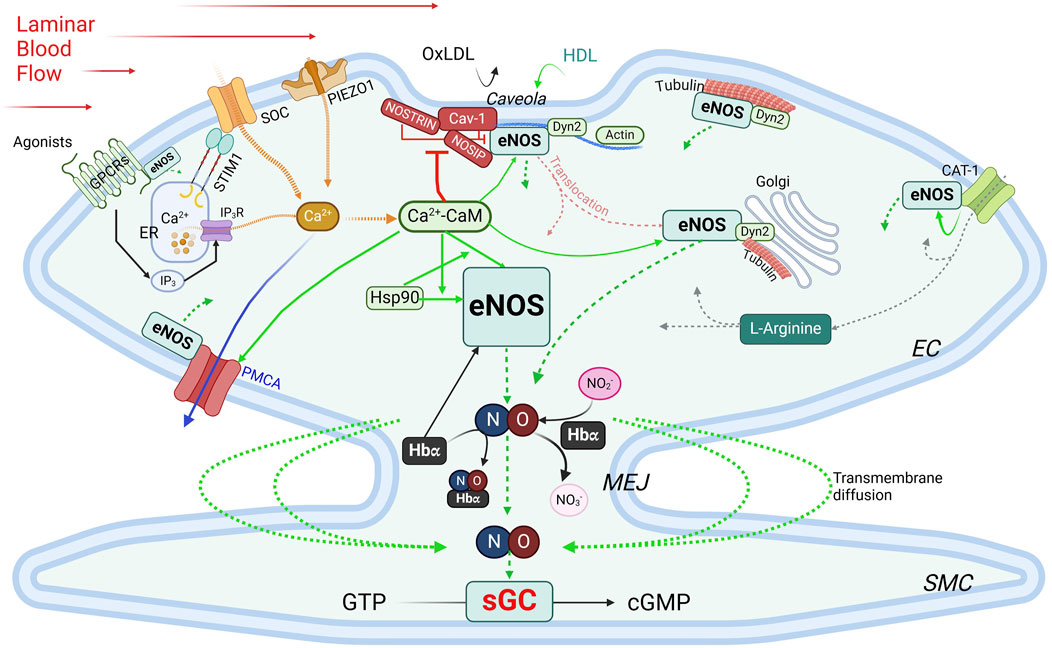
Figure 4. Regulation of eNOS by PPIs. Green boxes, stimulatory interacting partners; red boxes, inhibitory interacting partners. Green arrows indicate stimulatory interactions; dotted green arrows, NO production/diffusion; yellow dotted arrows, increase of intracellular Ca2+; solid blue arrow, Ca2+ extrusion; grey dotted arrows, L-Arginine transportation. See text for details. CAT-1, cationic anion transporter 1; Cav-1, caveolin-1; Ca2+-CaM, calcium-bound calmodulin; Dyn2, dynamin-2; EC, endothelial cells; ER, endoplasmic reticulum; GPCR, G protein-coupled receptor; Hbα, α subunit of hemoglobin; HDL, high-density lipoprotein; Hsp90, heat shock protein 90; IP3, inositol trisphosphate; IP3R, inositol trisphosphate receptor; MEJ, myoendothelial junctions; NOSIP, eNOS-interacting protein; NOSTRIN, eNOS trafficking inducer; ox-LDL, oxidized low-density lipoprotein; PMCA, plasma membrane Ca2+-ATPase; SMC, smooth muscle cell; SOC, store-operated Ca2+ channel; STIM1, stromal interaction protein 1.
Caveolin-1 (Cav-1) is the main caveolin of caveolae in endothelial cells (Feron et al., 1996; Garcia-Cardena et al., 1996; Ju et al., 1997). Cav-1 binds to eNOS at the caveolin-scaffolding domain (CSD, a.a. 81–101), preventing eNOS interaction with activators, thus inhibiting NO production under basal conditions (Garcia-Cardena et al., 1997; Michel et al., 1997). Physiological stimuli such as shear stress or G protein-coupled receptor (GPCR) agonists like bradykinin promote Ca2+ entry, leading to dissociation of the Cav-1-eNOS complex and allowing eNOS to be activated via phosphorylation by kinases such as Akt (Feron et al., 1998). Cav-1 deletion enhances endothelium-dependent relaxation and lowers blood pressure (Murata et al., 2007; Razani et al., 2001). Interaction with Cav-1, however, avoids excessive or aberrant eNOS activity, and Cav-1 deficiency can cause pulmonary hypertension and cardiomyopathy (Drab et al., 2001; Zhao et al., 2002). While plasma membrane localization is ideal for eNOS activation, this also exposes eNOS to external factors like ox-LDL and high-density lipoprotein (HDL). Ox-LDL can disrupt the cholesterol-rich environment in caveolae, thereby reducing NO production specifically from plasma membrane-bound eNOS, whereas Golgi-localized eNOS is more resistant (Zhang et al., 2006; Blair et al., 1999). HDL, on the other hand, provides cholesterol esters to maintain caveolae’s cholesterol, and via palmitoylation, retain eNOS at the plasma membrane (Uittenbogaard et al., 2000).
Ca2+-calmodulin eNOS activity is regulated by a complex and tightly regulated network of functionally and physically interacting proteins involved in Ca2+ signaling, including Ca2+ entry, the Ca2+ sensor CaM, and Ca2+ efflux. CaM, in its Ca2⁺-bound form (Ca2+-CaM), is required for activating eNOS by facilitating electron flow from the reductase domain to the oxygenase domain and promoting dimerization of the latter (Forstermann et al., 1991; Busse and Mulsch, 1990; Hellermann and Solomonson, 1997). The Ca2+ entry mechanisms in ECs play important roles in this process, as evidenced by the observations that removal of extracellular Ca2+ or inhibition of CaM suppresses agonist-induced NO production (Forstermann et al., 1991; Busse and Mulsch, 1990; Singer and Peach, 1982). Two major Ca2+entry pathways are important for NO production: store-operated Ca2+ entry (SOCE), the main agonist-induced Ca2+ entry mechanism in ECs (Abdullaev et al., 2008; Tran, 2020), and mechanosensitive Ca2+ entry, stimulated by blood shear stress. For activation of SOCE, the stromal interaction molecule 1 (STIM1) is required (Roos et al., 2005; Soboloff et al., 2012). Vascular STIM1 plays opposing roles in the regulation of vascular tone; smooth muscle cell STIM1 is important for VSMC contractility, proliferation and the development of hypertension (Kassan et al., 2016). On the other hand, endothelial STIM1 plays a critical role in the activation of eNOS to produce NO, such that EC-specific deletion of the STIM1 gene impairs endothelium-dependent vasorelaxation and increases blood pressure (Nishimoto et al., 2018). Shear stress, a potent physiological stimulus of NO production, stimulates PIEZO1 mechanosensitive channel for Ca2+ entry (Wang et al., 2016; Ranade et al., 2014; Li J. et al., 2014). Once bound to Ca2+, CaM regulates eNOS activity via two important mechanisms. First, the Ca2+-CaM complex displaces eNOS from the inhibitory interaction with caveolin (Michel et al., 1997). Second, CaM binds eNOS at a canonical CaM-binding site encompassing amino acids 493–512 (Venema et al., 1996) with a Kd value of ∼0.2 nM (Tran et al., 2005). This interaction is inhibited at low Ca2+ concentration by the autoinhibitory domain (residues 595–639) (Chen and Wu, 2000). Upon increases in intracellular Ca2+, Ca2+-bound CaM binds eNOS, displacing the autoinhibitory loop and facilitating electron transfer between the two domains (Pollock et al., 1991; Nishida and Ortiz de Montellano, 1999). Phosphorylation at Ser615 within this loop reduces the concentration of Ca2+ required for eNOS-CaM interaction, alleviating the autoinhibitory effect (Tran et al., 2008). CaM binding also enhances eNOS phosphorylation at Ser1177, which, in combination with Ser615 phosphorylation, further increases the Ca2+ sensitivity of eNOS-CaM interaction and synthase activation (Fleming et al., 2001; Tran et al., 2009; Tsukahara et al., 1994; Fleming et al., 1997). These phosphorylation events facilitate significant eNOS-CaM interaction and synthase activity at basal level of intracellular Ca2+ and explain the effects of factors that promote NO production without triggering significant increases in global cytoplasmic Ca2+. Interaction with Ca2+efflux channel – The plasma membrane Ca2+-ATPase (PMCA) is a key Ca2+ extrusion mechanism in ECs (Wang et al., 2002; Tran et al., 2003). eNOS directly interacts via residues 735 – 934 in its reductase domain with residues 428–651 in the catalytic domain of PMCA (Holton et al., 2010). This interaction enhances phosphorylation of Thr495 in the CaM-binding domain of eNOS (Holton et al., 2010), which reduces eNOS-CaM interaction (Fleming et al., 2001). The Ca2+/CaM-dependent phosphatase calcineurin is associated with the PMCA-eNOS complex, suggesting a potential role in the effect of PMCA expression on Thr495 phosphorylation status (Holton et al., 2010). Interestingly, PMCA activity is controlled by CaM interaction (Di Leva et al., 2008) and Ca2+ extrusion via PMCA moderates eNOS-CaM binding and synthase activation (Tran et al., 2016). Thus, CaM binding fine-tunes NO synthesis in response to subtle changes in the Ca2+ concentration surrounding eNOS and by control the activities of many of its interacting partners. With sub-nanomolar affinity for its interaction with CaM and limited abundance of CaM in ECs, eNOS expression level and activation in turn significantly influences the CaM-binding proteome in ECs (Tran et al., 2005; Tran et al., 2003). Treatment with 17β-estradiol or an agonist of the G protein-coupled estrogen receptor enhances CaM expression level substantially in ECs and promotes eNOS activity by moderating Ca2+ entry and efflux, enhancing eNOS-CaM interaction and associated eNOS phosphorylation (Tran, 2020; Tran et al., 2016; Fredette et al., 2017; Tran et al., 2015; Terry et al., 2017).
Interactions with components of the cytoskeleton and membrane-targeted proteins Actin interacts with an eight-amino acid motif (Jo et al., 2011; Higashi et al., 2002; Maier et al., 2000; Setoguchi et al., 2001; Fukuda et al., 2002; Holowatz and Kenney, 2011; Nystrom et al., 2004; Worthley et al., 2007) in the oxygenase domain of eNOS (Kondrikov et al., 2010). In in vitro assays, G-actin promotes eNOS activity more than does F actin (Su et al., 2003); however, a low G/F actin ratio appears to correlate with higher eNOS expression level (Searles et al., 2004). Dynamin-2 (Dyn2) is a GTP-binding protein in the caveolae and the Golgi (McClure and Robinson, 1996). Dynamin-2 interacts directly with eNOS in these compartments in ECs; this interaction is enhanced by Ca2+ and increases eNOS activity (Cao et al., 2001). NOSTRIN (eNOS trafficking inducer), a 506-a.a. protein enriched in vascular tissues, interacts with eNOS via an SH3 domain, promotes eNOS redistribution from the membrane, and inhibits synthase activity (Zimmermann et al., 2002). It also interacts with a region spanning a.a. 1- 61 of cav-1, N-terminally from the cav-1 scaffolding domain, thus forming a ternary complex with eNOS and cav-1 (Schilling et al., 2006). NOSTRIN interacts with dynamin-2 and is required for recruitment of eNOS to dynamin-containing structures (Icking et al., 2005). NOSIP (eNOS-interacting protein) is another protein residing in caveolae that interacts with eNOS and promotes its translocation from the plasma membrane and inhibit NO production (Dedio et al., 2001). In addition, eNOS associates with tubulin (Dedio et al., 2001), which plays an important role in its trafficking to the Golgi. Acetylation of α tubulin is involved in stabilizing microtubules where eNOS is associated in the Golgi and is phosphorylated for basal activity (Giustiniani et al., 2009). Interaction with GPCRs – Evidence from in vitro studies indicates that eNOS can interact with the juxtamembranous regions of AT1R, ETA, ETB and bradykinin B2 receptor (Marrero et al., 1999). While these interactions are likely important because activation of these receptors increases intracellular Ca2+, which is predicted to activate eNOS, they require further verification in vivo. Activity of eNOS is also regulated by its direct association with the cationic amino acid transporter (CAT)-1, the key transporter of L-arginine (Li et al., 2005). The promotion of eNOS activity by its interaction with CAT-1 is independent of L-arginine transport and is associated with enhanced phosphorylation at Ser1177 and Ser633 and reduced interaction with cav-1 (Li et al., 2005).
Heat shock protein 90 (Hsp90) is a molecular chaperone that stabilizes eNOS, thereby promoting its activation. The substrate-binding region of Hsp90 binds to the oxygenase domain of eNOS between a.a. 310–323 (Fontana et al., 2002; Xu H. et al., 2007). This interaction is increased by stimuli such as VEGF, histamine, estrogen, and shear stress (Garcia-Cardena et al., 1998; Russell et al., 2000; Venema et al., 1997). Hsp90 binding enhances phosphorylation at Ser1177 by recruiting kinases such as Akt, which further boosts NO production (Fontana et al., 2002). Hsp90 is critical for eNOS activity, and is involved in a reciprocal interactive relationship with eNOS and CaM: Hsp90 enhances both the magnitude and sensitivity of eNOS activation in response to Ca2+-CaM; in turn, the effect of Hsp90 to promote eNOS activity is enhanced by Ca2+-CaM complex (Takahashi and Mendelsohn, 2003). Hsp90 also exerts a CaM-independent effect to promote eNOS activity (Takahashi and Mendelsohn, 2003).
Hemoglobin alpha (Hbα) The α, but not β, subunit of hemoglobin is expressed in ECs in resistance blood vessels, where it is concentrated in the myoendothelial junctions (MEJs) between ECs and VSMCs (Straub et al., 2012). Hbα regulates NO availability via three mechanisms. First, it can bind and release NO and thus can act as both a reservoir and scavenger of NO, thereby regulating NO-mediated processes (Straub et al., 2014a; Keller et al., 2022; Straub et al., 2014b). In its ferrous (Fe2+) form, Hbα binds NO with high affinity, yielding nitrate and ferric (Fe3+) Hbα, which binds NO with much lower affinity (Straub et al., 2012). The reduction of Fe3+ Hbα to Fe2+ Hbα, catalyzed by cytochrome b5 reductase 3 (CytB5R3), thus enhances the scavenging action of NO by Hbα (Straub et al., 2012). Second, Hbα associates at its residues 34–43 with the oxygenase domain of eNOS in a macromolecular complex (Straub et al., 2014a), an interaction dictated by amino acids 36SFPT39 in the Hbα sequence (Keller et al., 2022). However, excessive NO scavenging in conditions of elevated Hb concentrations in the MEJs can reduce NO bioavailability and induce endothelial dysfunction (Denton et al., 2021), increasing vascular resistance (Straub et al., 2014a). Third, endothelial Hbα can function as a nitrite reductase, which produces NO through reduction of nitrite in hypoxic condition (Keller et al., 2022). These dynamic interactions help ensure that NO is available when needed, particularly in response to conditions like hypoxia and exercise, where vasodilation and increased tissue oxygen delivery are critical (Kim-Shapiro et al., 2005; Umbrello et al., 2013). In this context, it is worth noting that myoglobin is expressed in VSMCs and contributes to nitrite-dependent generation of NO in response to hypoxia independently of eNOS or iNOS (Totzeck et al., 2012).
Regulation of eNOS by substrates, co-factors, and product recyclingThe availability of substrates, co-factors, and the efficiency of product recycling are critical factors that dictate the outcome of eNOS activation. Disruptions in substrate availability, co-factor balance, or recycling processes—due to oxidative stress, metabolic disorders, or nutrient deficiencies—can impair eNOS activity and reduce NO bioavailability (Figure 5).
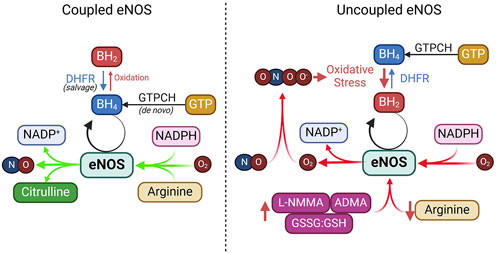
Figure 5. Coupled vs. uncoupled eNOS. See text for details. ADMA, asymmetric dimethylarginine; DHFR, dihydrofolate reductase; GTPCH, GTP cyclohydrolase I; GSSG, glutathione disulfide; GSH, glutathione; L-NMMA, NG-Monomethyl-L-arginine; NADPH, nicotinamide adenine dinucleotide phosphate.
L-arginine is the primary substrate for eNOS and is converted into NO and L-citrulline during the enzymatic reaction. Given its high affinity for eNOS (Km ∼2–3 μM) and an intracellular concentration above 100 μM, L-arginine is considered abundant in endothelial cells (Bode-Boger et al., 2007; Hardy and May, 2002). Nevertheless, extracellular L-arginine at millimolar concentrations can stimulate NO production (McDonald et al., 1997a; McDonald et al., 1997b). This has been considered the “arginine paradox.” Several mechanisms have been proposed to explain this phenomenon. One hypothesis involves the close association of eNOS with arginine transporters like CAT-1 and L-arginine recycling enzymes such as argininosuccinate lyase (ASL) (Erez et al., 2011). Other factors like competition from other amino acids, reduced uptake by cationic amino acid transporters (CATs), and elevated arginase activity—common in conditions such as hypertension and diabetes—can limit L-arginine availability to eNOS, diverting it away from NO production (McDonald et al., 1997a; McDonald et al., 1997b). Additionally, asymmetric dimethylarginine (ADMA), an endogenous inhibitor of eNOS, competes with L-arginine for binding, reducing NO synthesis (Boger et al., 1998a; Boger et al., 1998b). In addition to reducing NO synthesis, ADMA removes the suppression by NO of T-type voltage-dependent Ca2+ channels in VSMCs (Smith et al., 2020), triggering depolarizing Ca2+ spikes in VSMCs leading to vasoconstriction (Ng et al., 2024).
Tetrahydrobiopterin (BH4) is an essential cofactor for eNOS required for efficient electron transfer during the synthase’s catalytic cycle (Alp and Channon, 2004; Crabtree et al., 2009a; Bendall et al., 2014). BH4 keeps eNOS in a “coupled” state, in which it synthesizes NO rather than harmful superoxide (Alp and Channon, 2004; Crabtree et al., 2009b). BH4 can be produced through two pathways: de novo synthesis, regulated by the rate-limiting enzyme guanosine triphosphate cyclohydrolase I (GTPCH), or through the salvage pathway, where dihydrobiopterin (BH2) is recycled back to BH4 by dihydrofolate reductase (DHFR) (Crabtree et al., 2009b; Crabtree and Channon, 2011). Under oxidative stress, BH4 is rapidly oxidized into BH2 by superoxide anions or peroxynitrite, which is particularly strong during NO scavenging (Milstien and Katusic, 1999). This oxidative depletion of BH4 causes uncoupling of eNOS, where it produces superoxide instead of NO.
NADPH is a critical electron donor in eNOS and facilitates electron transfer through FAD and FMN for effective NO production. It also maintains the redox state of BH4, ensuring efficient NO synthesis (Reyes et al., 2015). Depletion of NADPH, as seen in glucose-6-phosphate dehydrogenase deficiency or due to excessive consumption by NADPH oxidases, leads to eNOS uncoupling and oxidative stress (Noreng et al., 2022). Additionally, activation of CD38 in post-ischemic heart injury can severely deplete NADPH, impairing eNOS function and disrupting NADPH-dependent BH4 salvage and synthesis (Reyes et al., 2015).
FAD and FMN, both derived from riboflavin (vitamin B2), are essential co-factors for eNOS’s electron transfer process. These flavins facilitate the flow of electrons from NADPH to the oxygenase domain, ensuring efficient NO production (Figure 1). Deficiency in FAD and FMN due to poor dietary intake or metabolic disorders can impair eNOS activity, resulting in reduced NO synthesis and increased oxidative stress.
eNOS uncouplingUnder certain conditions, eNOS can become “uncoupled,” shifting from producing NO to generating superoxide (O2⋅⁻), which not only reduces NO availability but also increases oxidative stress, reducing endothelial dysfunction. This section briefly explores the main causes of eNOS uncoupling and their therapeutic implications (Figure 5).
BH4 deficiencySuboptimal levels of BH4, or a decreased BH4/BH2 ratio, are significant contributors to eNOS uncoupling observed in hypertension, diabetes, and atherosclerosis (Vasquez-Vivar et al., 1998; Wever et al., 1997; Soltis and Cassis, 1991; Hong et al., 2001; Landmesser et al., 2003; Lee et al., 2009). Studies in animal models, such as hypertensive mice and rats and apolipoprotein E-deficient mice, have demonstrated that oxidative depletion of BH4 leads to increased eNOS-derived superoxide, which impairs vasorelaxation (Hong et al., 2001; Landmesser et al., 2003; Laursen et al., 2001). In humans, decreased BH4 levels and eNOS uncoupling have been linked to coronary artery disease, diabetes, and hypertension (Antoniades et al., 2007; Ismaeel et al., 2020; Heitzer et al., 2000b; Stroes et al., 1997). This imbalance is further exacerbated by oxidative stress, which directly oxidizes BH4 to BH2, reducing BH4 availability for NO synthesis (Vasquez-Vivar et al., 1998; Laursen et al., 2001; Guzik et al., 2002; Alp et al., 2003; Bendall et al., 2005). Additionally, reduced expression of GTP-cyclohydrolase 1, the rate-limiting enzyme in BH4 synthesis, and dihydrofolate reductase, the enzyme that recycles BH2 to BH4, further contributes to eNOS uncoupling in diabetes and hypercholesterolemia (Alp and Channon, 2004; Xu et al., 2009; Xu J. et al., 2007; Whitsett et al., 2007; Munzel and Daiber, 2017; Chuaiphichai et al., 2017).
L-arginine deficiency or imbalanceL-arginine depletion is a significant contributor to eNOS uncoupling. Although intracellular L-arginine concentration typically far exceeds its Km for eNOS, obesity, diabetes, and cardiovascular diseases can cause L-arginine deficiency. In such cases, reduced availability of L-arginine limits its ability to act as a substrate for eNOS, favoring the production of ROS over that of NO (Bode-Boger et al., 2007; Yang and Ming, 2013). A key mechanism of L-arginine depletion is upregulation of arginase, an enzyme that competes with eNOS for L-arginine and converts it into urea and L-ornithine. Arginase induction is particularly prevalent in obesity, diabetes, and atherosclerosis (Lass et al., 2002). For example, oxidized LDL significantly increases ARG2 expression levels in ECs (∼20%) but increases its activity by ∼80% (Ryoo et al., 2006). Many other factors can upregulate the expression level and activity of arginase, such as lipopolysaccharide, TNF-α, glucose, thrombin, hypoxia and angiotensin II [reviewed in Pernow and Jung (2013)]. Inhibiting arginase or supplementing L-arginine can restore NO production and improve endothelial function in these conditions (Chicoine et al., 2004). Furthermore, imbalance between L-arginine and ADMA, an endogenous inhibitor of eNOS, leading to reduced L-arginine/ADMA ratio, contributes to eNOS uncoupling (Closs et al., 2012; Watson et al., 2016; Peyton et al., 2018).
Oxidative stress and reactive oxygen species (ROS)Oxidative stress plays a pivotal role in disrupting endothelial function by promoting eNOS uncoupling. Excessive ROS overwhelms the antioxidant defense system and causes oxidative stress that damages lipids, proteins, and DNA, contributing to the development of CVD (Thannickal and Fanburg, 2000; Costa et al., 2021; Sharifi-Rad et al., 2020; Li H. et al., 2013). Risk factors such as dyslipidemia, diabetes, hypertension, obesity, and smoking increase ROS levels, increasing endothelial dysfunction and CVD progression (Cai and Harrison, 2000; Xiang et al., 2021; Jurcau and Ardelean, 2022). ROS also play a key role in ischemia-reperfusion injury (Xiang et al., 2021; Jurcau and Ardelean, 2022). In the vascular wall, enzymes such as NADPH oxidase, xanthine oxidase, and uncoupled eNOS generate ROS, particularly superoxide. Superoxide reacts with NO to form peroxynitrite (ONOO−), a toxic compound that depletes NO and worsens endothelial dysfunction (Li H. et al., 2014; Thomson et al., 1995; Victor et al., 2009). NADPH oxidases, particularly NOX2, are major sources of superoxide in diabetes, hypertension, smoking, and aging (Konior et al., 2014; Gray et al., 2013; Manea et al., 2018; Fukui et al., 1997; Marchi et al., 2016; Kim et al., 2014; Jiang et al., 2011). The oxidative depletion of BH4 by ROS, especially via NADPH oxidases, further exacerbates eNOS uncoupling (Konior et al., 2014; Zhang et al., 2020).
eNOS glutathionylationS-glutathionylation at Cys689 and Cys908 in the reductase domain results in eNOS uncoupling (Figure 3) (Chen et al., 2010; Chen et al., 2013; Suvorava et al., 2015; Zweier et al., 2011). This modification typically occurs under oxidative stress, when the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) decreases (Suvorava et al., 2017; Benson et al., 2013; Daiber et al., 2002). S-glutathionylation disrupts the alignment of FAD and FMN, essential cofactors for eNOS’s electron transport, resulting in superoxide production instead of NO (Chen et al., 2010; Zweier et al., 2011). This mechanism of eNOS uncoupling is unique in that it occurs in the reductase domain, unlike other uncoupling mechanisms that occur in the oxygenase domain, and can be inhibited by L-NAME (Chen et al., 2010). In vivo studies have confirmed the association of eNOS S-glutathionylation with endothelial dysfunction in hypertension, aging, and cardiovascular diseases (Chen et al., 2010; Suvorava et al., 2015). In ex vivo rat aortic segments and spontaneously hypertensive rats, high levels of eNOS S-glutathionylation correspond with impaired vasodilation. S-glutathionylation can be reversed by Grx-1 (Chen et al., 2013; Shang et al., 2017).
Strategies to enhance endogenous no productionThe use of exogenous NO donors is limited by the short-lived nature of their effects and the development of nitrate tolerance. Enhancing endogenous NO production should provide physiological and more sustained effects. Current strategies involve promoting eNOS activity by providing essential substrates or cofactors or by pharmacologically upregulating eNOS expression and activity; this could also be done by preventing/reversing eNOS uncoupling. We will divide our review below of these approaches based on the level of available evidence.
Approaches with both preclinical and clinical evidenceL-arginine supplementationRationaleL-arginine supplementation to promote NO production is based on the premise that in conditions of endothelial dysfunction, intracellular L-arginine levels may become suboptimal. It is also based on the “arginine paradox,” discussed above, that despite an intracellular L-arginine concentration much higher than its Km for eNOS activity, high extracellular concentration of L-arginine can still promote NO production. Additionally, L-arginine also competes with ADMA, an endogenous eNOS inhibitor (Boger et al., 1998c), thereby restoring the L-arginine/ADMA ratio that is critical for eNOS activity (Dong et al., 2011) (Figure 6).
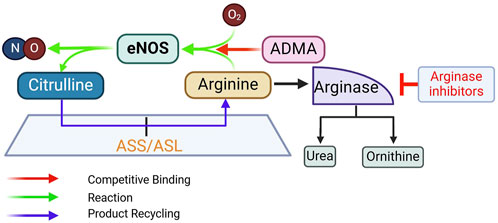
Figure 6. Rationale for supplementing L-arginine, inhibiting arginase, and supplementing L-citrulline to promote eNOS activity. ADMA, asymmetric dimethylarginine; ASL, argininosuccinate lyase; ASS, argininosuccinate synthase. See text for more details.
EvidenceEarly preclinical studies provided evidence that oral L-arginine supplementation improves acetylcholine (ACh)-induced vasorelaxation and NO production (Girerd et al., 1990; Cooke et al., 1991; Rossitch et al., 1991; Boger et al., 1995). Clinical studies began with intravascular injection of L-arginine in small groups of subjects. In hypercholesterolemic patients (n = 8, mean age 51.5) with slight luminal irregularities of the left anterior descending coronary artery (LAD), ACh-induced reduction in coronary blood flow, an indication of endothelial dysfunction, is improved by intracoronary injection of L-arginine (Drexler et al., 1991). In patients with diffuse atherosclerotic LADs (n = 13, age 56 ± 7.5), direct intracoronary L-arginine injection also ameliorates the ACh-induced vasoconstriction and reduction in blood flow (Dubois-Rande et al., 1992). In patients with critical peripheral limb ischemia (n = 10, age 68.3 ± 3.1), a single intravenous dose of L-arginine significantly increases blood flow, accompanied by urinary cGMP excretion (Bode-Boger et al., 1996). Overall, intravascular L-arginine delivery in humans offers acute improvement of endothelial function. In young healthy subjects (n = 80, age 25.4 ± 0.2), this effect is achieved with higher doses and is correlated with L-arginine plasma concentrations (Bode-Boger et al., 1998).
From clinical trials testing the effects of oral administration of L-arginine, a picture has emerged that short-term L-arginine administration (≤3 months) provides benefits, whereas longer supplementation (≥6 months) gives mixed results. For example, 4-week L-arginine supplementation improves reactive hyperemia in patients with hypertension and hyperhomocysteinemia (2.4 g/d, n = 25, age 40–65) (Reule et al., 2017); and flow-mediated dilation is improved by 3-month L-arginine supplementation in hypertensive subjects (2.4 g/d, n = 40, age 40–65) (Menzel et al., 2018). In patients with a history of coronary bypass surgery, 6-month supplementation with L-arginine (6.4 g/d, n = 32, age 65 ± 10) reduces ADMA levels, increases plasma cGMP, and improves reactive hyperemia compared to placebo (n = 32, age 64 ± 11) (Lucotti et al., 2009). However, 6-month supplementation with L-arginine in patients with peripheral arterial disease (3 g/d, n = 66, age 73 ± 9) does not increase NO synthesis or improve vascular reactivity vs. placebo (3 g/d, n = 67, age 72 ± 7) (Wilson et al., 2007) or alter vascular stiffness in post-myocardial infarction (MI) patients (3 × 3 g/day, n = 75, age 60.4 ± 12.9) (Schulman et al., 2006). This has led to the “arginine tolerance” hypothesis (Wilson et al., 2007), akin to the common nitrate tolerance phenomenon.
ChallengesGiven the state of current evidence, major cardiovascular guidelines do not include L-arginine supplementation for the prevention of CVD. Among explanations for the absence of effect of long-term L-arginine administration in some clinical trials, oxidative depletion of BH4 can result in eNOS uncoupling even with sufficient L-arginine supply (Xiong et al., 2014; Scalera et al., 2009). L-arginine also increases arginase expression, which in turn reduces L-arginine availability (Caldwell et al., 2018; Wang et al., 2006), a mechanism that may partly explain the arginine tolerance hypothesis. Nevertheless, interpretation of results of
留言 (0)