
記住我
Fasciolopsis buski, commonly known as the giant intestinal fluke, is one of the largest digenean trematode flatworms. This parasite causes fasciolopsiasis, which is prevalent in South Asian countries, including the Indian Subcontinent. It is important for veterinary and medical care because it affects both humans and pigs frequently. Dogs and rabbits can also contract the disease, but pigs serve as reservoirs. Severe infestation can be fatal if untreated, causing significant intestinal damage such as ulceration, erosion, abscesses, and haemorrhage (Biswal et al., 2013; Biswal et al., 2016a; 2018; Roberts et al., 2009). Notably, about 60% of infected individuals in India and mainland China remain asymptomatic.
F. buski thrives in regions where pig breeding is practiced and the consumption of aquatic plants like water chestnut, lotus, caltrop, and bamboo is prevalent. In India, this parasite is the sole documented species of its genus, prevalent in warm, moist areas such as the north-eastern part of India and exhibits morphological variation across geographic isolates (Roy and Tandon, 1993; Ridha et al., 2021; Hsu, 1964; Rajendran et al., 2019). The highest incidence rates are found in children, who typically ingest plants that have encysted metacercariae. The intestinal mucosa develops ulceration lesions and localised inflammation as a result of the parasite. There have been reports of an edematous state caused by protein-losing enteropathy in cases of severe infection (Sen-Hai and Mott, 1994).
The best way to control fasciolopsiasis is to avoid eating uncooked, water-based foods. Cooking aquatic vegetation or temporarily submerging plants or nuts in boiling water is typically enough to prevent infection. Also, restricting the use of human faeces as fertiliser and promoting modern pig rearing can help prevent fasciolopsiasis. This approach has been supported with large-scale screening protocols and improvements to water, sanitation and hygiene. Successful health education initiatives have reduced the transmission of this parasite in some endemic locations, such as Taiwan; but, in areas where fasciolopsiasis was assumed to be eliminated, such as Uttar Pradesh, in India, re-emerging infections has been observed (Bhatti et al., 2000).
Fasciolopsiasis is now managed in a way similar to other trematode diseases, with the goal of preventing transmission between mammalian hosts, including humans, and intermediate gastropod hosts, such as Lymnaea species. Praziquantel (PZQ) is a popular medicine due to its excellent efficacy in cases with fasciolopsiasis (Ridha et al., 2021; Taraschewski et al., 1986). Despite the efficiency of existing medications, the efficacy of vaccinations against helminth parasites like trematodes is mainly unknown. Recently, there has been growing concern regarding the development of anthelmintic resistance in fasciolopsiasis. Even though it has not become widespread, the appearance of resistance has led to a growing interest in other control/therapeutic methods, such as vaccines. At the time of writing, no vaccination has undergone human clinical trials or been authorised for commercial use in pigs.
Developing a vaccine for parasites like F. buski is therefore crucial due to rising drug resistance and the scarcity of new drugs (Nakagawa, 1922; Mas-coma et al., 2005). F. buski, like most helminths, has a complex eukaryotic multicellular life cycle, which complicates vaccination (Mas-coma et al., 2005; Sharma et al., 2015). The development of a vaccine would contribute to the WHO 2030 Roadmap for NTD goal of eliminating fasciolopsiasis as a public health problem (Malecela and Ducker, 2021).
While significant progress has been made in discovering possible vaccine molecules to reduce fasciolosis and fasciolopsiasis in livestock, it is reasonable to argue that the level of efficacy needed to bring the product to market has not yet been attained. Till date, three methods have been employed to find potential vaccine candidates against F. hepatica and F. gigantica: (i) examining antigens that exhibit cross-reactivity with sera from animals infected with other trematode infections; (ii) identifying orthologous antigens that have been identified as potential vaccine candidates in other species; and (iii) selecting antigens rationally that are crucial for liver fluke survival (Perera and Ndao, 2021). Many vaccine trials in livestock have assessed the efficacy of these potential antigens, including some single or chimeric experimental F. hepatica vaccines (Cox, 2002). Fatty acid binding proteins (FABP), glutathione S-transferase (GST), cathepsin L1 (CatL1), peroxiredoxin (Prx), and the gut-associated exopeptidase leucine aminopeptidase (LAP) have all been examined as potential leads. Similar studies on the intestinal fluke F. buski are lacking, and there is a need to design multi-subunit vaccines shared by F. hepatica and F. gigantica, which could lead to the development of a cost-effective vaccine with efficacy against both liver and intestinal flukes.
Understanding the functional role that each parasite molecule plays at each stage of the life cycle is crucial for developing new control methods. Over the last decade or so, the development of sequencing technologies and their application to the study of Fasciola species has paved the way for a global molecular understanding of the developmental biology of parasites. Comprehensive data on genomics, transcriptomics, proteomics, and glycomics has provided us with an in-depth knowledge of the parasite life cycle, from DNA synthesis and expression to the manufacture and post-translational modification of numerous essential parasite proteins. This understanding can be leveraged to build innovative control approaches. The availability of substantial data on the transcriptomes of intestinal flukes (F. buski) can help with this approach, as well as the identification of new vaccine targets (Biswal et al., 2018). It is important to note that mathematical modelling indicates that fasciolid vaccines, even if they do not provide complete protection, could significantly contribute to fluke control while also delaying the spread of anthelmintic resistance through reduced use of triclabendazole (Turner et al., 2016).
The aim of this study was to create and test a fasciolopsiasis vaccine for the first time utilising an in silico approach (Figure 1). This method has already been utilised to create vaccines against different pathogens for a variety of diseases, including SARS CoV-239 (Singh et al., 2020), Mycobacerium TB (Bibi et al., 2021), Schistosoma mansoni (Sanches et al., 2021), F. hepatica, F. gigantica, and Onchocerca volvulus (Shey et al., 2019). Previously, we report on the entire transcriptome and draft genome study of F. buski, which aided us in characterising some of its key biological properties and provided vast resources for the creation of a viable diagnostic system and anti-parasitic treatment strategies. In order to better combat these multicellular organisms, we have categorised these proteins into key biological processes (such as proteases, protease inhibitors, antioxidants, and immunomodulators) (Biswal et al., 2018) that can be combined to create a cocktail vaccine utilising the antigens found in F. buski to prevent infection in several hosts and obstruct the zoonotic transmission of the intestinal fluke. This could provide a way to get around the molecular redundancy and block or interfere with key parasitic processes.

Figure 1. Schematic illustration of the workflow using B cell, CTL, and HTL epitopes followed by molecular docking, molecular dynamics simulation, and in silico cloning.
2 Materials and methods2.1 Selection and retrieval of vaccination targetsA fundamental understanding of the parasite biology has been achieved through the transcriptome data of the adult stage of this giant intestinal fluke, which was previously reported with Bioproject Accession: PRJNA212796 ID: 212,796, and is archived in the NCBI SRA: SRP028107, SRX327895, SRX326786, SRX316736, as well as in the North-East India Helminth Parasite Information Database (NEIHPID) developed by our group (Biswal et al., 2016b). This information is highly informative for the identification of potential drug targets and host-pathogen interaction studies.
The transcriptome of F. buski contains significant metabolic pathways, including glycan biosynthesis, lipid metabolism, carbohydrate metabolism, energy metabolism, amino acid metabolism, and nucleotide metabolism. The analysis of these pathways encompassed all genes related to glycolysis, the Krebs cycle, and fatty acid metabolism. The annotated genes were compared to those available in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using BLASTx. The mapped genes illustrated metabolic pathways involving key biomolecules, including carbohydrates, amino acids, and various other pathways. The glycolytic pathway includes all the essential enzymes, including hexokinase, enolase, pyruvate kinase, and lactate dehydrogenase.
Pathway analysis is an approach for identifying vaccination targets by analysing the activity of confirmed gene sets that are biologically related. This strategy may be more effective since it focusses on gene sets rather than individual gene expression levels. The major components of metabolic pathways discovered in the F. buski transcriptome are depicted in Figure 2. Coloured edges indicate proteins that are homologous to any F. buski unigene. The skewed representation of enzymes in the pathway analysis have revealed insights into gene regulation and parasite biology, suggesting to a catabolic process and a possible reliance on their host for nourishment at several stages of their life cycle. However, pathway analysis has certain drawbacks, such as inadequate pathway information and the inability to include non-protein coding elements. Therefore, we relied on the combined approach of target proteins from previous studies (Williams et al., 2013; Smooker et al., 2010; Wijffels et al., 1994; Marcilla et al., 2008; Vicente et al., 2016; Chaithirayanon et al., 2006; Capron et al., 2001; Ehsan et al., 2021) and tallied that with pathway analysis from the transcriptome data manually. Some of the key proteins used in design of the chimeric vaccine viz. Thioredoxin glutathione reductase (Amino acid metabolism), Cathepsin B (Energy metabolism), Cathepsin L (Glycolysis/gluconeogenesis), Leucine amino peptidase (Amino acid metabolism), Fatty acid-binding protein type 3 (Fatty acid metabolism), 14-3-3 protein epsilon (Energy metabolism), Glutathione S-transferase (xenobiotics biodegradation and metabolism; Glutathione metabolism) and Tegumental calcium-binding EF-hand protein 3 & 4 (Lipid metabolism) are all indicated in Figure 2. The “names, “cellular locations,” and “functions” of each of the nine proteins used in the multi-subunit vaccine design and the rationale behind the choice of these vaccine targets are summarized in Table 1. A comprehensive description of these proteins is provided below.”
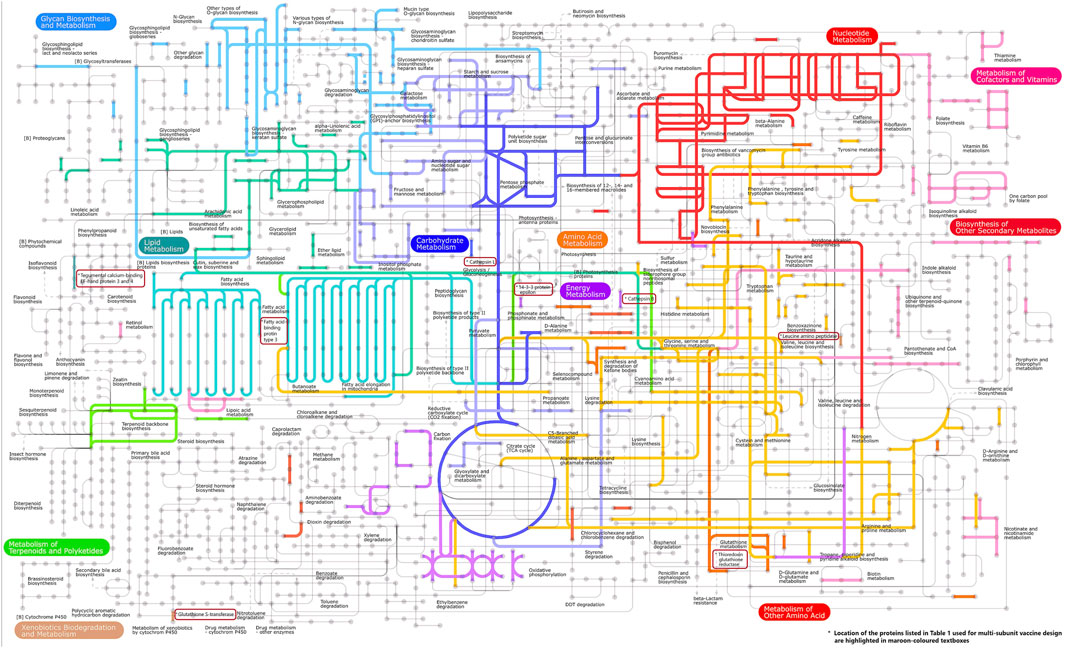
Figure 2. Important components of the metabolic pathways found in the transcriptome of F. buski. Colored edges indicate proteins that are homologous to any F. buski unigene. The positions of several key proteins mentioned in Table 1, which are utilized in the design of chimeric vaccines, are highlighted in maroon-colored textboxes. These proteins include Thioredoxin glutathione reductase (related to amino acid metabolism), Cathepsin B (associated with energy metabolism), Cathepsin L (involved in glycolysis/gluconeogenesis), Leucine amino peptidase (linked to amino acid metabolism), Fatty acid-binding protein type 3 (related to fatty acid metabolism), 14-3-3 protein epsilon (associated with energy metabolism), Glutathione S-transferase (involved in xenobiotics biodegradation and metabolism; Glutathione metabolism), and Tegumental calcium-binding EF-hand proteins 3 and 4 (related to lipid metabolism).
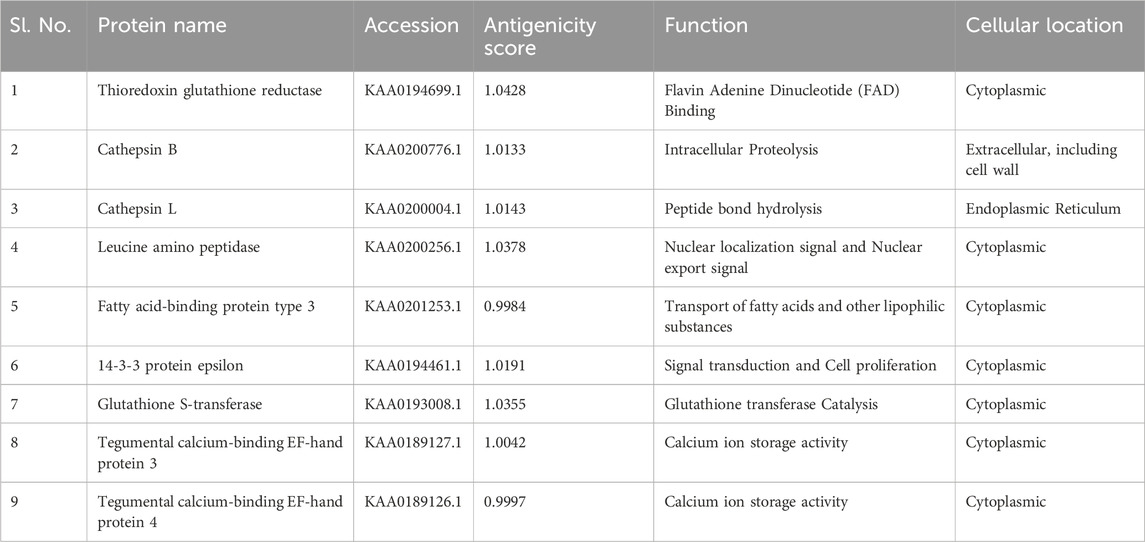
Table 1. List of proteins used for multi-subunit vaccine design showing their NCBI accessions and antigenicity Score.
Rationale behind the choice of proteins listed in Table 1 as vaccine targets.
A total of 9 different genes and respective proteins were selected (KAA0194699.1, KAA0200776.1, KAA0200004.1, KAA0200256.1, KAA0201253.1, KAA0194461.1, KAA0193008.1, KAA0189127.1, and KAA0189126.1) and obtained from NCBI in FASTA format. Additionally, the structure of TLR2 (PDB ID: 5D3I) was sourced from NCBI. The antigenicity of all proteins was predicted on the online server (http://imed.med.ucm.es/Tools/antigenic.pl).
2.1.1 Thioredoxin glutathione reductaseA single enzyme, thioredoxin glutathione reductase (TGR), which is a fusion of a glutaredoxin (Grx) domain to classical thioredoxin reductase (TR) domains, provides electrons to oxidized glutathione (GSSG) and thioredoxin (Trx) in the simplified thiol-based redox system of Platyhelminth parasites. The use of TGR as a schistosomiasis medication target has been confirmed (Williams et al., 2013).
2.1.2 Cathepsin BTrematode (or fluke) cathepsin B proteases are widely distributed in juvenile and immature flukes. Cathepsin Bs are important for trematode biology, according to recent studies, mostly in Fasciola, that use RNA interference (RNAi), inhibitors, and vaccinations (Chantree et al., 2013). The damage caused by flukes when they infiltrate host tissues will be much reduced if these proteases are rendered inactive by chemotherapy or immunization, since they are primarily produced by infectious parasite stages. Cathepsin Bs are thus confirmed to be important strategic targets for fluke control (Smooker et al., 2010).
2.1.3 Cathepsin LCathepsin L is a cysteine protease superfamily member that is extensively distributed in parasitic species. It plays critical roles in worm invasion, migration, food intake, moulting, and immunological evasion. Studies using pure native FheCL1 and FheCL2 as vaccines in sheep and cattle have demonstrated that these enzymes can produce strong anti-embryonation/hatch rate effects that would prevent the spread of parasites, as well as protection, ranging from 33% to 79%, against experimental challenges involving F. hepatica metacercariae (Wijffels et al., 1994).
2.1.4 Leucine amino peptidaseThe enzymes leucine aminopeptidase (LAP) and phosphoenolpyruvate carboxykinase have been identified by researchers as immunodominant antigens detected by sera from patients with fasciolosis using proteomics and immunoblotting techniques. Prior findings indicate that LAP elicits a robust and specific reaction, indicating its possible application in the serologic diagnosis of human F. hepatica infections (Marcilla et al., 2008).
2.1.5 Fatty acid-binding protein type 3Fatty-acid-binding proteins are among the substances that have been linked to interfering with the host response (FABPs). Approximately 15 kDa in size, FABPs are a multigenic family of small cytosolic proteins that primarily bind hydrophobic fatty acid ligands noncovalently. They exhibit distinct tissue distribution patterns and are linked to numerous important biological processes, such as the control of cellular lipid homeostasis, cell growth and differentiation, cellular signalling, gene transcription, and the immune response. In the gastrointestinal system of their hosts, helminth parasites thrive in an oxygen-poor environment where they are unable to synthesize most of their own lipids de novo, including cholesterol and long-chain fatty acids. Because helminths rely on the host’s fatty acids for intracellular lipid oxidation via FABP transportation, FABPs are essential to the biosynthesis of fatty acids and cholesterols in helminths. FABPs are an attractive choice as drug targets for novel anthelminthic vaccines because of their significant function in lipid oxidation, and their predominance in a variety of nematode, trematode, and cestode species (Vicente et al., 2016).
2.1.6 14-3-3 protein epsilonThe 14-3-3 protein family comprises tiny, acidic proteins that operate as scaffolding, chaperones, and adaptors to regulate many intracellular signaling pathways. These proteins are involved in numerous physiological processes that are currently being studied for pharmaceutical intervention. Tested as prospective vaccine candidates in mice, the 14-3-3 proteins of S. mansoni and Echinococcus multilocularis show promise for use as defense mechanisms (Chaithirayanon et al., 2006). Clearly, to fully utilize the promise of 14-3-3-proteins as therapeutic targets, additional fundamental understanding as well as enhanced models and pharmacological candidates are needed.
2.1.7 Glutathione S-transferaseEnzymes called glutathione S-transferases (GSTs) are found in parasites like trematodes and aid in the detoxification of foreign compounds in cells. GST is a target antigen that has been identified and molecularly cloned which has allowed for the demonstration of its vaccine potential in a number of animal species, including rodents, cattle, and primates, as well as the consistent demonstration of vaccination’s ability to decrease female worm fecundity and egg viability through the production of neutralizing antibodies (IgA and IgG). S. haematobium GST, Sh28GST, has been used in phase I and II clinical trials after encouraging preclinical research (Capron et al., 2001).
2.1.8 Tegumental calcium-binding EF-hand protein 3 & 4Tegumental proteins (TegPs) are members of the calcium-binding protein (CaBP) family, which also includes a dynein light chain-like domain at the C-terminus and two EF-hand motifs at the N-terminal domain. Numerous helminth parasites, such as S. mansoni, E. multilocularis, Schistosoma hepatica, Schistosoma gigantica, Clonorchis sinensis, and Opisthorchis viverrini, have been found to have CaBPs. CaBPs have been categorised into approximately 13 isoforms in S. mansoni and 4 isoforms in F. hepatica and F. gigantica (CaBP-1–4). Studies on the biochemical aspects of CaBPs from other parasites have revealed that, although the structural models of all family members are remarkably similar, these proteins’ dimerization, ion-binding, and drug-binding properties differ. Their crucial function in immune processes during host-parasite interactions may be demonstrated by this (Ehsan et al., 2021).
2.2 Epitope predictionThe B-cell epitopes were predicted using ABCPred tools (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html). The B-cell epitope database (BCIPEP) and an artificial neural network (ANN) were used for this (Saha and Raghava, 2006). The helper T-lymphocyte (HTL) epitopes were identified using MHC-II tools from the immune epitope database (IEDB, http://tools.iedb.org/mhcii/). Except for the alleles HLA-DPA1*01:03/DPB1*02:01 and HLA-DRB1*07:01, which encompass 99.9% of the global population [22,23], all parameters were left at their default values. Epitopes were chosen based on their lowest percentile rank and IC50. Cytotoxic T-lymphocyte (CTL) epitopes were predicted using the NetCTL 1.2 tools (http://www.cbs.dtu.dk/services/NetCTL/), a default threshold value of 0.75 and three allele supertypes (A2, A3, and B7) with a population coverage of 88.3% were configured in the tools. IFN-γ inducing epitope prediction was used to identify positive and negative inducers (http://crdd.osdd.net/raghava/ifnepitope/predict.php) (Dhanda et al., 2013). The parameters were maintained at their default settings, with the exception of the Motif and SVM hybrid, as well as IFN-γ versus other cytokine model.
2.3 Toxicity and epitope identity assessmentThe ToxinPred module (http://crdd.osdd.net/raghava/toxinpred/multi.submit.php) was used to determine the toxicity of the chosen epitopes and show whether they were toxic or not. Standard parameters like Sensitivity, Specificity, Accuracy, and Matthew’s correlation coefficient (MCC) were taken into consideration while assessing the performance of the models. Default physiochemical properties (Hydrophobicity, Hydrophilicity Charge and Molecular Weight) were considered for predicting the toxicity of the epitopes used in this study. All parameters have been set to their default values (Gupta et al., 2013). Furthermore, epitopes were tested for identity in humans and pigs. These epitopes were run through a BLASTp (Basic Local Alignment Search Tool protein) search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (Altschul et al., 1990) and were cross-checked using the Multiple Peptide Match tool in PIR (https://research.bioinformatics.udel.edu/peptidematch/batchpeptidematch.jsp) (Wu et al., 2003). Epitopes were matched against the human and pig proteomes using the UniProt/SwissProt option, which included the various isoforms. Epitopes anticipated to be harmful or identified in humans or pigs were excluded, and a new epitope was chosen for testing.
2.4 Vaccine designThe vaccine was designed by using the adjuvant followed by B-cell, CTL and HTL epitopes joined by KK, AAY and GPGPG linkers respectively. Linkers were employed to arrange the amino-acid into favourable conformations. An Adjuvant is employed to enhance the immunogenicity of the corresponding vaccine.
2.5 Antigenicity, allergenicity and physiochemical parametersThe capacity to bind B-cell or T-cell receptors is referred to as antigenicity. Tools ANTIGENpro and VaxiJen (Doytchinova and Flower, 2007a; Doytchinova and Flower, 2007b) were used to assess the vaccine’s antigenicity. The Allertop (https://www.ddg-pharmfac.net/AllerTOP/) (Dimitrov et al., 2014) and Algpred (https://webs.iiitd.edu.in/raghava/algpred/submission.html) (Saha and Raghava, 2006) servers were used to evaluate the allergenicity of the vaccine design. Models of bacteria and parasites were employed with the VaxiJen v2.0 server, and a 0.7 threshold was applied in each case. Using the ToxinPred site and the previously described approach, the toxicity of vaccines was assessed. It is anticipated that the multi-epitope vaccination will be non-toxic, non-allergic, and antigenic. With the Expasy ProtParam tool (https://web.expasy.org/protparam/) (Wilkins et al., 1999), the construct’s physicochemical parameters were examined. Molecular weight, in vitro and in vivo half-lives, instability index, theoretical isoelectric point (pI), aliphatic index, and grand average of hydropathicity (GRAVY) were among the attributes that were analysed. The vaccine solubility upon overexpression in E. coli was assessed with SOLPro (http://scrat.ch.proteomics.ics.uci.edu/) (Magnan et al., 2009). The tools PSIPRED 4.0 (http://bioinf.cs.ucl.ac.uk/psipred/) (Buchan and Jones, 2019) and RaptorX Property Prediction (https://github.com/j3xugit/RaptorX-3DModeling) (Wang et al., 2016) were used to predict the vaccine secondary structure (2D).
2.6 Prediction of structure of the vaccine, refinement, validation and dockingThe I-Tasser server (http://raptorx.uchicago.edu/StructurePrediction/predict/) was used to predict the construct vaccine’s tertiary structure (Källberg et al., 2012). ProsA was used for structural validation, and Pdbsum was used to create the Ramachandran plot (Wiederstein and Sippl, 2007; Laskowski et al., 2018). The model was further optimised using the Galaxy server (Heo et al., 2013), and RMSD was used to determine which model was the best. TLR2 (PDBID: 5D3I) was obtained from the PDB. ClusPro (http://nrc.bu.edu/cluster) was used for docking, and among the generated models, the most optimal model was chosen based on binding energy for the vaccine construct (Kozakov et al., 2013; Kozakov et al., 2017). Further the Staphylococcal superantigen-like protein 3 (SSL3) complex with TLR was excluded because it interacts with the TLR2 complex through lipopeptides and blocks the binding site of the TLR2 ligand, interfering with the formation of heterodimers between TLR2 and TLR1. Numerous publications have detailed the ligand-induced interactions of TLR2 with other pattern recognition receptors, including integrins, scavenger receptors, CD14, and a variety of other receptors, all of which are crucial for TLR2 activity. These “information-driven” approaches are successful in TLR2-complex docking carried out by ClusPro because of the conserved aspects of the binding interface. Of all the models generated from ClusPro, the most effective model in terms of binding energy to bind the vaccine construct was chosen. The binding residue information was supplied as attractive residues. These information in the TLR2, following residues that are involved in SSL3 binding viz. ASP294, TYR326, LEU328, SER329, TYR332, SER354, ASP294 and ASP327, Phe349, Leu350, Gln375, Tyr376, Asn379, Phe349, Leu350, Gln375, Tyr376, and Asn379, His358, Leu324, Tyr326 were taken based on previously published experimental data. To specify attraction, we entered the residues that are believed to participate in the binding into one or both sides of the interface. In the docking calculations, an extra attractive force was applied to the selected residues. The following whole adjuvant sequence (LprA) was inserted in vaccine construct as shown below (LprA being agonist of TLR2).
(MKHPPCSVVAAATAILAVVLAIGGCSTEGDAGKASDTAATASNGDAAMLLKQATDAMRKVTGMHVRLAVTG
DVPNLRVTKLEGDISNTPQTVATGSATLLVGNKSEDAKFVYVDGHLYSDLGQPGTYTDFGNGASIY
NVSVLLDPNKGLANLLANLKDASVAGSQQADGVATTKITGNSSADDIATLAGSRLTSEDVKTVPTTVWI
ASDGSSHLVQIQIAPTKDTSVTLTMSDWGKQVTATKPV).
The molecular dynamics simulation (MDS) for the vaccine was performed on Ubuntu 2020.1 gromacs version. The docked complex was immersed in a TIP3P water model and the AMBER99SB force field. The Genion tool was employed to add 7 Cl− to achieve system neutralization and to mitigate steric clashes of the system they were kept in energy minimization and force below 1,000 kj mol−1 nm−1. Long-range electrostatics were calculated using the Particle Mesh Ewald (PME) method, while Lennard-Jones and coulomb were computed with a cut-off distance of 1.0 nm. Bond lengths were constrained using the LINCS algorithm, and the SHAKE algorithm was employed to find out water bond. Following energy minimization, the system was equilibrated at 1 ns of NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature), and the system was run for 100 ns. Analysis tools g_rms, g_rmsf, and g_hbonds were used to calculate the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and number of hydrogen bonds, respectively (Abraham et al., 2015).
2.8 In-silico cloningCodon optimisation is critical for performing in silico cloning in a bacterial expression system. This procedure was carried out using the Java Codon Adaptation Tool (http://www.jcat.de/), with the E. coli K12 strain as the host organism. The parameters selected were “avoid Rho independent terminators,” “avoid prokaryotic ribosome binding sites,” and “avoid restriction enzyme cleavage sites.” The optimal Codon Adaptation Index (CAI) and GC concentration are 0.8%–1.0% and 30%–70%, respectively. The optimised vaccination sequence was then reversed to include XhoI and BamHI restriction sites at the N and C terminals. The integration of the sequence was assisted using the pET28a(+) vector (d'Aubenton Carafa et al., 1990; Carbone et al., 2003; Grote et al., 2005).
2.9 Immune simulation for vaccine efficacyIn silico simulations of the human immune response to the chimeric molecule were conducted using the C-IMMSIM web server (https://wwwold.iac.rm.cnr.it/∼filippo/c-immsim/index.html). We administered three doses, 4 weeks apart. To mimic 1,050 simulation steps, injections of 1,000 vaccine proteins were administered 4 weeks apart at 1, 84, and 168 time-steps (each time-step is equivalent to 8 h in real life). The remaining parameters were kept at their defaults (Greenwood, 2014).
3 Results3.1 Selected vaccination targets and respective epitopes for vaccine designWe have focussed on chimeric molecule vaccine development against fasciolopsis by considering those molecules which are present at the host–parasite interface, specifically proteins within the parasite’s tegument and gut, extracellular vesicles, which appear in the parasite’s secretome and play a critical role in host invasion, host immune modulation/manipulation, and parasite survival. Transcriptomic and proteomic studies have increased our understanding of these important proteins by unmasking their expression and secretory profiles in the various F. buski developmental life cycle stages. The antigenicity all the nine sequences for F. buski as shown in Table 1, was assessed based on the antigenic score (Table 1) to boost the immunogenicity of the protein TLR-2 agonist Lipoprotein LprA (P9WK55) (Figure 1).
3.2 Anticipated immune properties of the chimeric molecule vaccinationThe ABCpred server was used to predict B-cell epitopes, and the top nine epitopes were chosen from nine proteins (Table 2). All protein sequences were then submitted to IEDB MHC-II for HTL epitope prediction. Nine epitopes were selected from the proteins based on their low percentile score and IC50 value. Notably, epitopes with high binding affinity had the lowest percentile values (Table 3). Nine epitopes were selected from the proteins based on their low percentile score and IC50 value. Notably, epitopes with high binding affinity had the lowest percentile values (Table 3). The parameters remained unchanged, with the exception of a selection of alleles, LA-DPA1*01:03/DPB1*02:01 and HLA-DRB1*07:01, based on the disease’s geographical distribution. The NetCTL1.2 server was utilised to identify the CTL epitopes. The vaccine design used the A2, A3, and B7 supertypes of the MHC class, out of 27. The default threshold of 0.75 was used, and the epitopes with the highest scores were picked, totalling twenty-seven (Table 4). The vaccine was ultimately built around three B-cell, three HTL, and nine CTL epitopes. All epitopes tested positive for IFN-γ production (Table 3). Furthermore, the toxicity of the epitopes was determined using the ToxinPred programme, which demonstrated that all epitopes were non-toxic (Table 5).
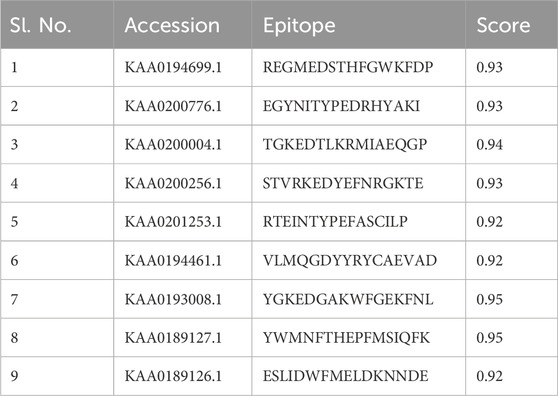
Table 2. List of selected B-cell epitopes based on highest scores for the proteins sequences used in this study.

Table 3. List of selected HTL epitopes for the proteins sequences under study and their IFN-γ inducing properties.
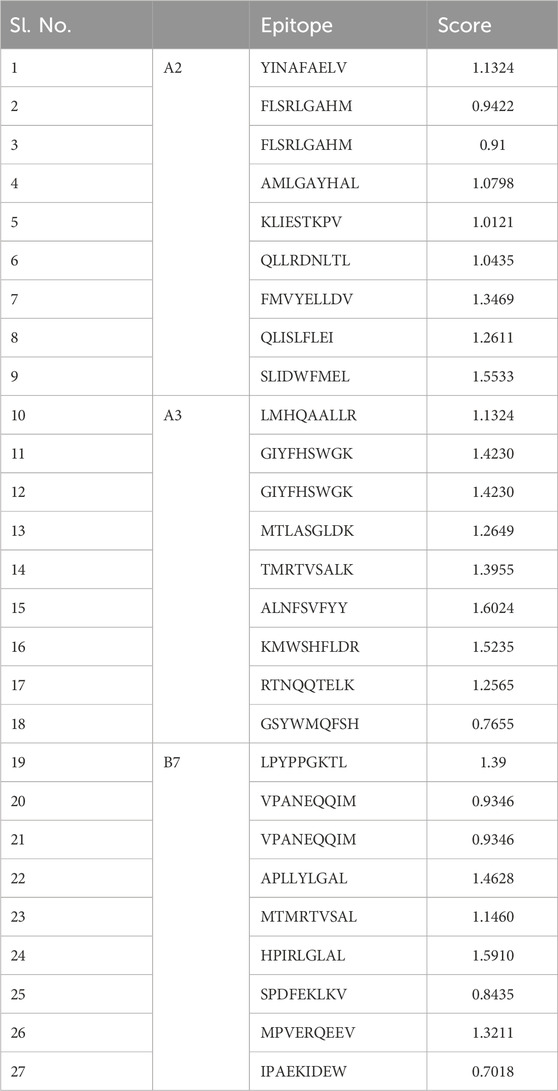
Table 4. List of selected CTL epitopes for proteins sequences used in this study.
Table 5. Toxicity of the epitopes was assessed using the ToxinPred tool.
3.3 Methodology for vaccine constructTo construct the vaccine, we designed the most favourable CTL, HTL, and B-Cell epitopes using AAY, GPGPG, and KK linkers, respectively. Furthermore, an EAAAK linker was used to bind Lipoprotein LprA Adjuvant (NCBI ID: P9WK55) to the vaccine’s the N-terminal. The vaccine’s structure comprises of nine HTL epitopes, twenty-seven CTL epitopes, and nine B-Cell epitopes derived from the target protein sequences. The use of a TLR-2 agonist, Lipoprotein LprA (P9WK55), increased the vaccine’s immunogenicity due to its adjuvant qualities. All of the identified epitopes from B-cell (9-mers), HTL (9-mers), and CTL (27-mers) were linked using particular linkers EAAAK between adjuvant and epitopes. After evaluating and comparing several structures, we determined the vaccine’s final structure with 901 amino acids. The final recombinant vaccine was analysed for subsequent evaluations.
3.4 Antigenicity, allergenicity and physiochemical propertiesThe predicted vaccine design had antigenicity values of 0.6244 and 0.5541 in the ANTIGENpro and Vaxijen servers, respectively. Allertop and Algpred determined the allergenicity of the construct vaccine, which indicated that it is non-allergenic. In the allergenicity testing utilising Allergen FP tools, the protein with the highest Tanimoto similarity index is uniprotkb Q2RB59 (0.85). ProtParam, an online programme, was used to predict the vaccine construct’s physiochemical properties. The molecular weight of the 901 amino acid protein was 98.71 kDa, and its theoretical PI was 8.74. The vaccine construct had an instability score of 27.36 and an aliphatic index of 76.93, which confirmed the protein’s thermostability. The GRAVY score of −0.136 indicates the protein’s hydrophilic nature. Therefore, the resulting vaccine is extremely acidic, thermostable, and hydrophilic, making it suitable to undergo subsequent efficacy testing.
3.5 Vaccine modelling and validationThe 3-D model of the vaccine construct was predicted using Raptor-X (Figure 3) and was subjected to additional refinement on the Galaxy server to acquire a valid structure. The Ramachandran plot derived from the model was 89.2%, 8.2%, 1.8%, and 0.8% in the most favoured regions (Supplementary Figure 1A), additional allowed region, generously allowed region, disallowed region. Following refinement, the validate structure exhibit 91.5% in most favoured regions, 7.6% in additional allowed region, 0.1% in generously allowed region, 0.5% in disallowed region (Supplementary Figure 1B). The ProsA server display (Z-score was −10.4) (Supplementary Figures 2A, B), indicating the qualitative characteristics of the vaccine construct. The energy plot indicated that the initial few sequences exhibit slightly elevated energy levels, followed by a substantial decrease, suggesting that the protein is thermodynamically stable (Supplementary Figure 2C). Protein Sol server was performed that showed deviation population average, charge per amino acid and fold propensity (Figure 4). Secondary structure was predicted by pdbsum server (Supplementary Figures 3A, B).
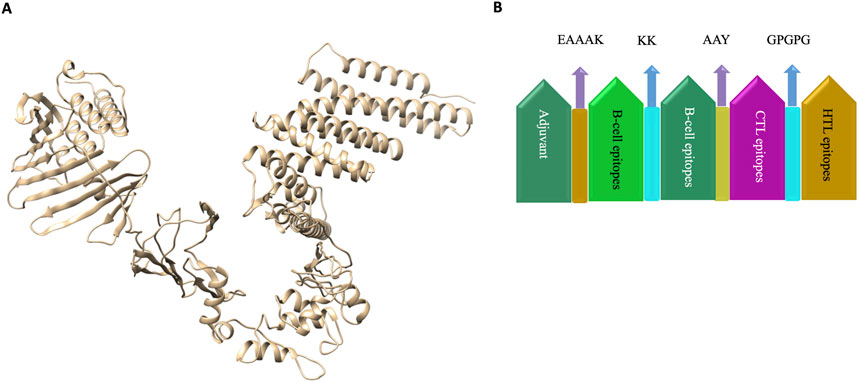
Figure 3. (A) 3D structure of vaccine construct. (B). Vaccine was designed by using the adjuvant followed by B-cell, CTL and HTL epitopes joined by KK, AAY and GPGPG linkers respectively. Linkers were employed to arrange the amino-acid into favourable conformations. An Adjuvant is employed to enhance the immunogenicity of the corresponding vaccine.
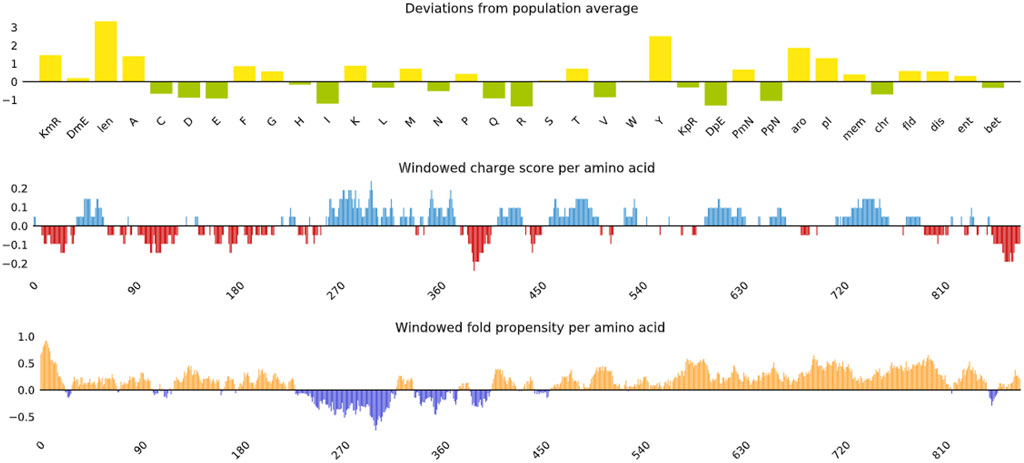
Figure 4. Vaccine construct solubility as determined by the protein sol server that showed deviation population average, charge per amino acid and fold propensity.
3.6 Molecular docking with vaccine and TLR2To investigate the relationship between the TLR2 (5D3I) and the vaccine construct, molecular docking was used. Out of the sixteen models that were generated, the optimal model for the dynamics investigation between TLR2 and vaccination (Figure 5) and interaction (Figure 6) was selected based on the lowest binding energy of −1,287.2 kJ.mol−1. For the complicated profiles of mobility (NMA B-factors), deformability, eigenvalues, covariance map, and linkage matrix, iMODS server was used (Figures 7A–D, 8A).
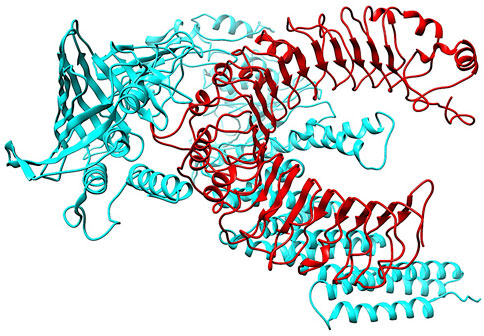
Figure 5. Molecular Docking of TLR2 and vaccine complex. To investigate the relationship between the TLR2 (5D3I) and the vaccine construct, molecular docking was used. Of the sixteen models that were generated, the optimal model for the dynamics investigation between TLR2 and vaccination and interaction was selected based on the lowest binding energy of −1,287.2 kJ.mol−1.

Figure 6. Interaction between TLR2 and the vaccine construct. The interaction map of the vaccine construct with chains A and B from TLR2. Hydrogen bonds are shown by dashed (green) lines with the length of the bond printed in the middle.
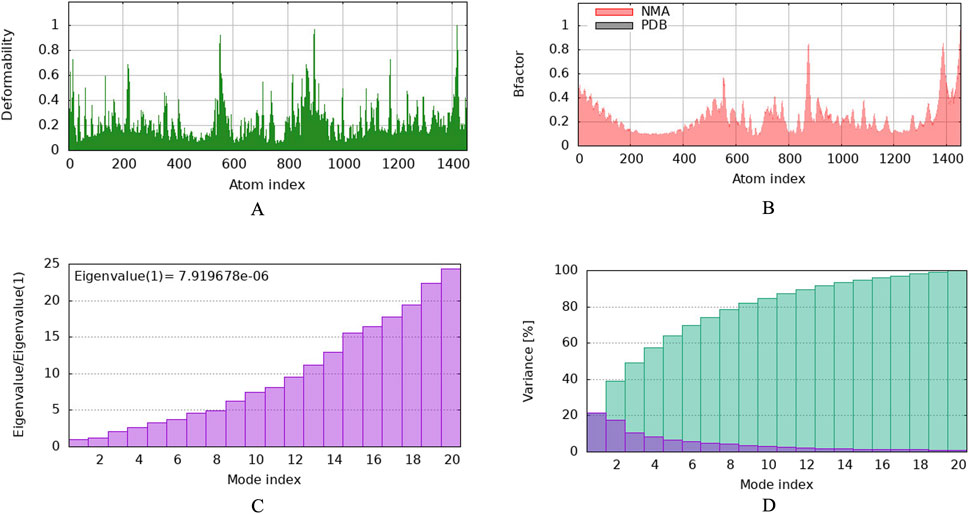
Figure 7. Molecular dynamics simulation of vaccine protein complex. Stability of the vaccine depicted via graphical representation. (A) Deformity vs. Atomic index of the vaccine construct, (B) Bfactor vs. Atomic index of the vaccine, (C) Eigenvalue vs. Atomic index of the vaccine construct, (D) Variance vs. Atomic index of the vaccine construct.
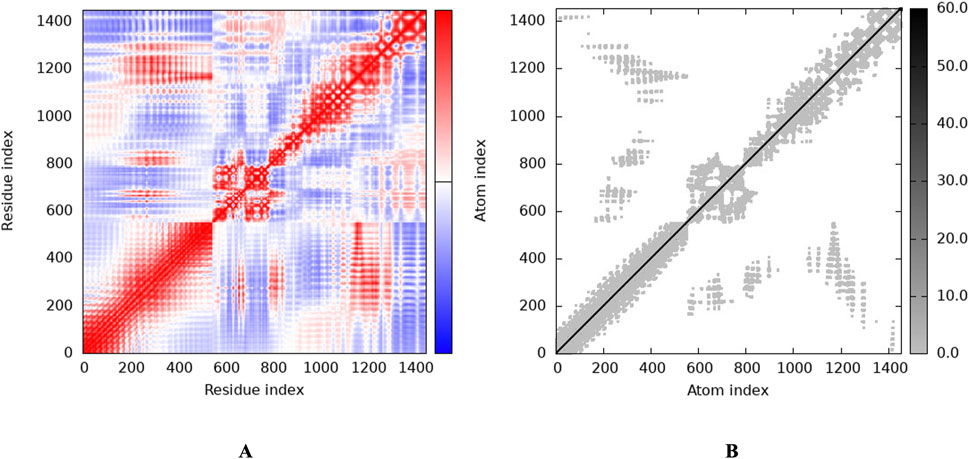
Figure 8. Stability of the vaccine via graphical representation. (A) Representation of the Residue index (covariance), (B) Representation of the Atomic index (elastic network analysis).
3.7 Dynamics study of the complexA molecular dynamics simulation (MDS) of the TLR2-vaccine complex was run for 150 ns to evaluate its thermodynamic properties. The parameters RMSD, RMSF, Rg, and hydrogen bonds were calculated. After 10 ns, the system reached stability. Analysis of Cα atoms revealed an average RMSD of 0.84 nm ± 0.15 (Figure 9A). The RMSF revealed residue variations, with an average value of 0.27 ± 0.15 nm (Figure 9B). The protein-protein complex’s stability is indicated by its hydrogen bonds, which averaged 963.67 ± 19.15 (Figure 9C). Rg values, which indicate protein compactness, averaged 4.17 ± 0.02 nm (Figure 9D).
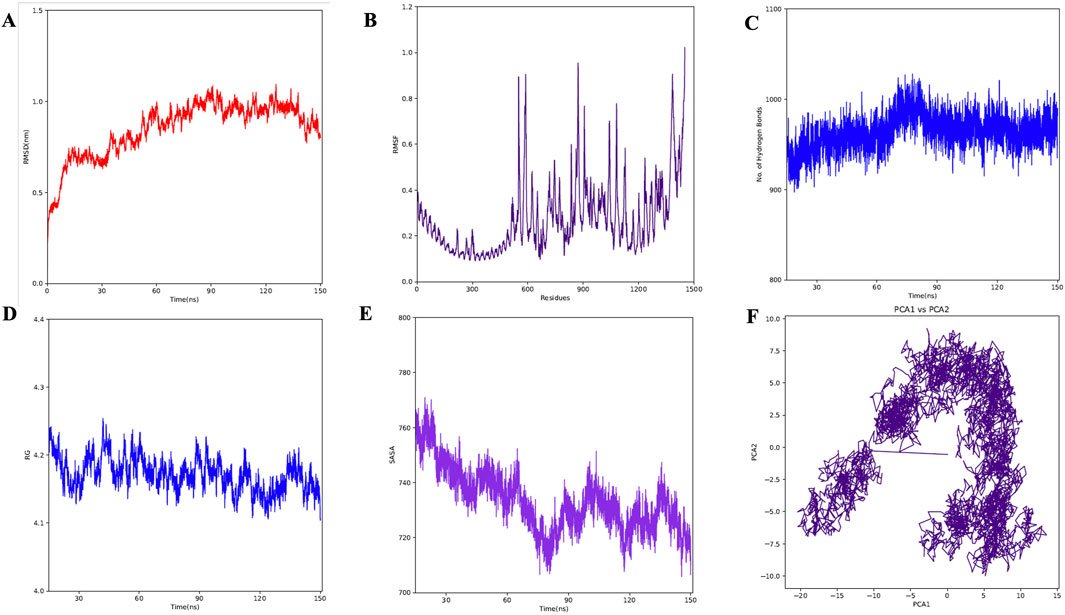
Figure 9. MD simulation of complex. (A) RMSD for the amino acid backbone of the complex, (B) RMSF of amino acids side chain of the complex (C) Number of hydrogen bonds formed during last 50 ns, (D) Radius gyration (Rg) of the complex, (E) Surface area (SASA) of the vaccine construct, (F) Principal Component Analysis (PCA) of the vaccine construct.
3.8 In-silico cloning into pET28 (+) vectorThe pET (Plasmid for Expression by T7 RNA Polymerase) system is renowned for producing large amounts of protein. Over 220,000 research articles have utilized this system, with more than 40,000 involving pET28a (+). Key advantages include: (i) Strong T7 Promoter: Increases protein production, (ii) Common Restriction Sites: Especially upstream of the T7 promoter, making it ideal for expression cloning, (iii) Lac Operator Regulation: Suppresses uninduced expression, (iv) Self Lac Repressor Encoding: Reduces promoter leakiness, especially beneficial for bacteria with lac repressor deficiencies and (v) Medium Copy Number (20–25 per cell): allows high expression levels without overloading the cell. The proposed vaccine design was cloned in silico in the E. coli vector backbone following codon optimisation in order to carry out the expression analysis. The codons in the vaccine design were modified to match those in the E. coli expression system using the JCat online tool (https://www.jcat.de/). On the Codon Adaptation Index (CAI), a perfect score of 1 was obtained. Similarly, 50.73 percent GC concentration was determined to be optimal for achieving high protein expressions. The pET28 (+) vector was used to clone the vaccine because of its appropriate CAI (1.0) and GC (50.73) content, which indicated a higher likelihood of expression in E. Coli cells. The N- and C-terminal portions of the vaccine construct were modified to include XhoI and BamHI restriction sites, as the original build did not have these sites. The cloned map (Figures 10A, B) was visualized and created using the SnapGene software (https://www.snapgene.com/). The resulting restriction c
留言 (0)