Mice
Mice were housed in standard cages under a 12-h light cycle and 12-h dark cycle (dark from 6:00 PM to 6:00 AM) at standard ambient temperature and humidity conditions and were provided with ad libitum water and a standard chow diet (Purina-LabDiet, Prolab RMH 3000). For the mouse studies, a minimum of 5 mice per experimental group was calculated using G*Power software. This was based on the ability to detect with 80% power a 20% difference in whole body and fat pad weight with a SD of 10% based on a 2-sample t-test with alpha = 0.05 (2-sided). Mice were assigned randomly to different types of diets without being screened for inclusion or exclusion by the investigators.
Ethics approval: All experiments were performed using a protocol approved by the Institutional Animal Care and Use Committee at Wake Forest University School of Medicine in facilities approved by the American Association for Accreditation of Laboratory Animal Care.
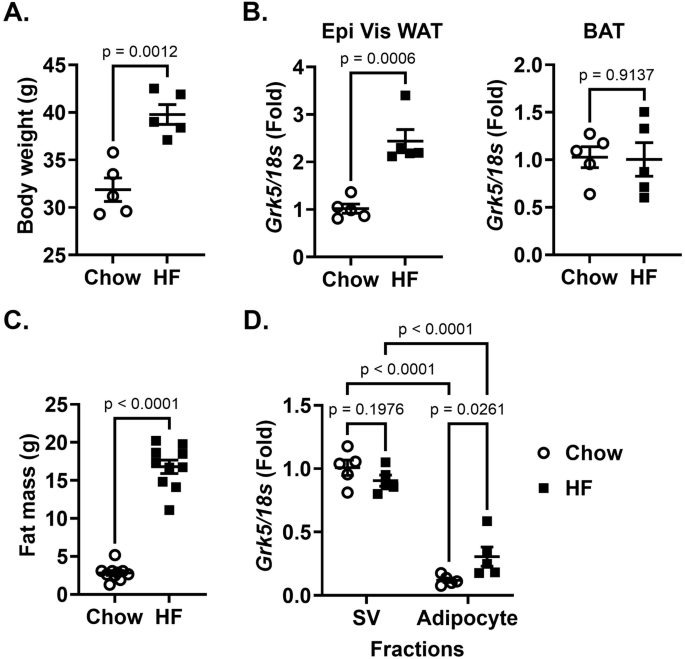
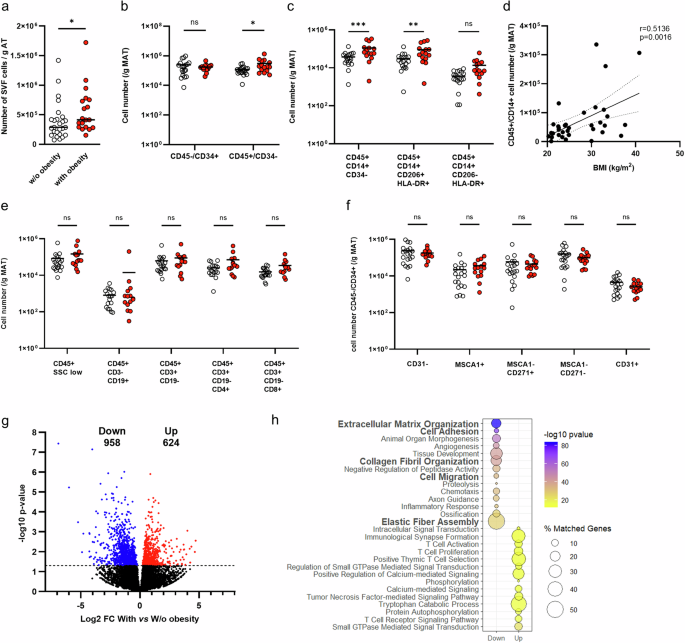
To assess the impact of high fat diet on Grk5 expression, 8-week-old male C57BL/6J mice (Jackson Lab, Bar Harbor, MN, USA, Strain #000664) were fed chow or a high fat diet (Envigo, Indianapolis, IN, USA, #TD 88137, 42% from fat, 0.2% total cholesterol or Research Diets Inc, New Brunswick, NJ, USA, #D12451, 45% from fat) for 16 weeks. Six-week-old female C57BL/6J mice were fed chow or a high fat diet (Research Diets Inc, New Brunswick, NJ, USA, #D12492, 60% from fat) for 12 weeks. Mice were fasted overnight before being euthanized, and adipose tissue (including visceral, subcutaneous, and brown) were collected and stored at −80 °C until used for gene or protein expression. To determine where in adipose tissue Grk5 is expressed, 6-week-old male C57BL/6J mice were fed chow or a high-fat diet (Research Diets Inc, New Brunswick, NJ, USA, #D12492, 60% from fat) for 12 weeks. After 10 weeks on diet, mice went through EchoMRI™ analysis, which measures body composition (e.g., fat mass) of live mice. After 12 weeks on diet, mice were fasted for 16 h and epidydimal visceral white fat pads were harvested and used for adipose tissue digestion as described in the section below.
Adipose tissue digestion
Briefly, adipose tissue was enzymatically digested in a digestion buffer (0.5 g of fat in 10 ml) containing 0.8 mg/ml of collagenase II (Worthington Biochemical Corp., Lakewood, NJ, USA), 3% of fatty acid free-BSA (Sigma-Aldrich, Burlington, MA, USA), 1.2 mM of calcium chloride (Sigma-Aldrich), 1 mM of magnesium chloride (Sigma-Aldrich), and 0.8 mM of zinc Chloride (Sigma-Aldrich) in Hanks Buffered Salt Solution (Life Technologies, Carlsbad, CA, USA) for 60 min in a shaking water bath at 37 °C with 200 rpm agitation. The fat digest was then filtered through a 250-um filter (Fisher Scientific, Pittsburgh, PA, USA). The adipocyte fraction and stromal vascular (SV) fraction were collected by centrifugation at 800 × g for 10 min. Red blood cells in the SV fraction were lysed using ACK lysis buffer. The adipocyte and SV fractions were treated with QIAzol Lysis Reagent (Qiagen, Venlo, Netherlands) and stored at −80 °C until used for gene expression.
Cell cultures
The 3T3-L1 preadipocyte cell line was purchased from ATCC (CL-173™). The GRK5 KO 3T3-L1 preadipocyte was generated using CRISPR gene editing and provided by Synthego Corporation, Redwood City, CA (cells are available upon request). The guide sequence (i.e., TATGTGACAAGCAACCAATT) was designed to target exon 3 of Grk5. The KO clone was cut and had a nucleotide removed (i.e., A) during the non-homologous end joining repair process, resulting in a frameshift mutation that causes premature termination of translation at a new nonsense codon, as confirmed by the Sanger sequence (Supplementary Fig. 2A).
GRK5 KO 3T3-L1 and its wildtype (WT) control 3T3-L1 preadipocytes were first used to assess their proliferation rate using Click-iT® EdU cell proliferation kit (ThermoFisher Scientific, Waltham, MA, USA) based on the manufacture’s procedure. Briefly, cells were seeded into a 6-well plate with a density of 0.1 × 106 per well in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Billings, MT, USA) supplemented with 10% iron-fortified calf serum (CS, Sigma-Aldrich) and 1% penicillin/streptomycin (P/S, Gibco) for 24 h. Cells were then treated with EdU (5-ethynyl-2’-deoxyuridine) solution and incubated for 24 h. EdU, a nucleoside analog of thymidine, was incorporated into newly synthesized DNA and fluorescently labeled with a bright, photostable Alexa Fluor™ 647 dye. Total DNA was stained using Hoechst 3342 and imaged using BioRad ZOE Fluorescent Cell Imaging System.
GRK5 KO 3T3-L1 and WT control 3T3-L1 preadipocytes were cultured and differentiated into adipocytes as described previously [20]. Briefly, preadipocytes were seeded at 0.05 × 106 cells per well in a 6-well culture plate. Cells were cultured in DMEM supplemented with 10% iron-fortified CS and 1% P/S for 48 h until ∼90% cell confluence. Adipogenesis (Day 0) was induced by changing the medium to DMEM containing 10% fetal bovine serum (FBS, Sigma-Aldrich) plus an adipogenic cocktail (Sigma-Aldrich) including 1 μg/ml of insulin, 0.25 μM of dexamethasone, 0.5 mM of 3-isobutyl-1-methylxanthine, and 2 μM of rosiglitazone for 3 days (Day 3). Cells were then treated with 1 μg/ml of insulin only for 3 days (Day 6) and then without any adipogenic reagents for the next 3 days (Day 9). The medium was changed every 2 days. At Days 0, 3, 6 and 9 of differentiation, cells were stained with Oil-Red O and imaged using the BioTek Cytation C10 Confocal Imaging Reader (Agilent BioTek, Winooski, VT, USA) as well as lipid extracted for triacylglycerol (TAG) measurement as previously described [20].
In order to examine ERK expression, Day 2 differentiated WT and GRK5 KO cell cultures were serum starved overnight and then treated with 1 μg/ml of insulin for 5, 10, and 15 min. The cellular proteins were harvested as described in the section below for Western blot analysis.
RNA extraction and real-time PCR
Total RNA was harvested from cells and tissues using QIAzol Lysis Reagent and isolated by following the protocol described in the RNeasy Lipid Tissue Mini Kit (Qiagen). The concentration and quality of RNA were determined using a Nanodrop (ThermoFisher Scientific) and standardized to 1 μg of RNA for cDNA synthesis. The cDNA was prepared with the OmniScript RT Kit (Qiagen) and stored at −20 °C until used for real-time PCR. Real-time PCR was performed in duplicate on the QuantStudio™ 3 systems (ThermoFisher Scientific) using TaqMan® Fast Advanced Master Mix and TaqMan® gene expression assays (ThermoFisher Scientific) including Grk5 (Mm00517039_m1), Cd36 (Mm00432403_m1), Fabp4 (Mm00445878_m1), Pparγ (Mm0040940_m1), Acc1 (Mm01304257_m1), Fasn (Mm00662319_m1), Dgat2 (Mm00499536_m1), Lipin1 (Mm00550511_m1), and Lipin2 (Mm00522390_m1) with 18S rRNA (REF 4352655) as a housekeeping gene. Gene expression was normalized to the endogenous control gene 18S rRNA (REF 4352655) and analyzed using the 2ddCt method with 95% confidence.
Protein extraction and Western blot
Total cellular protein was harvested in Pierce™ IP lysis buffer (ThermoFisher Scientific) supplemented with cOmplete™ EDTA-free Protease (Sigma-Aldrich) and PhosSTOP™ Phosphatase (Sigma-Aldrich) inhibitor tablets and frozen at −20 °C until used. Protein samples were normalized to 1 mg of protein, prepared in non-reducing laemmli buffer and DTT, and heated at 95 °C for 10 min. Protein was loaded and separated on a 4–20% polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA, USA) and transferred to a 0.2 μm nitrocellulose membrane (Bio-Rad). Membranes were blocked in 5% non-fat milk in 1X Tris-buffered saline plus 0.1% Tween (TBST, Bio-Rad) for 2 h at room temperature. Primary antibodies were diluted in TBST with 1% non-fat dry milk and incubated overnight at 4 °C with gentle rocking. Primary antibodies were diluted as follows: GRK5 (#sc-518005, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:1000, GRK2 (#74761, ThermoFisher Scientific) at 1:1000, GAPDH (#sc-137179, Santa Cruz Biotechnology) at 1:1000, PPARγ (#2435, Cell Signaling Technology, Danvers, MA, USA) at 1:1000, CD36 (#28109, Cell Signaling Technology) at 1:1000, α-tubulin (#2144, Cell Signaling Technology) at 1:1000, Phospho-p44/42 ERK1/2 (#4376, Cell Signaling Technology) at 1:1000 and Total p44/42 ERK1/2 (#4695, Cell Signaling Technology) at 1:1000. Following overnight incubation, membranes were washed 3 times in TBST for 5 min with agitation and incubated with secondary antibody in 5% non-fat milk for 1 h at room temperature (ThermoFisher Scientific mouse and rabbit secondaries, 1:5000) with gentle rocking. Membranes were washed 3 times in TBST for 5 min with agitation. SuperSignal™ West Pico PLUS Chemiluminescent Substrate (ThermoFisher Scientific) was added to the membrane prior to imaging using the ChemiDoc Gel Imaging System (Bio-Rad). Protein expression was quantified using Bio-Rad ImageLab software.
RNA sequencing and pathway analysis
GRK5 KO and WT cells were seeded and proliferated for 48 h. After 48 h, cells were treated with the adipogenic cocktail as described above for 6 h, and RNA was collected and extracted as previously described [20]. Total RNA was used to prepare cDNA libraries using the Illumina® TruSeq Stranded Total RNA with Ribo-Zero Gold Preparation kit (Illumina Inc., San Diego, CA, USA). The libraries were pooled and sequenced to an estimated target read depth of 40 M single-end 100 bp reads per sample on the Illumina NovaSeq 6000. For all samples, 80% of sequences achieved >Q30 Phred quality scores (FASTQC analysis, Babraham Bioinformatics). Adapter contamination was cleaned with Trimmomatic [21]. Reads were aligned to the murine reference genome mm39 using the STAR sequence aligner [22], and gene counts determined using featureCounts software [23]. Differentially expressed genes were identified using limma [24]. We then used Ingenuity Pathway Analysis (IPA) to identify top up-and down-regulated pathways.
Computational molecular modeling and in vitro inhibition assays of a GRK5 inhibitor
A pyridine-based bicyclic compound of small molecule GRK5 inhibitor, GRK5-IN-2, was purchased from MedChemExpress (HY-136561). The binding affinity of GRK5-IN-2 to GRK5 and GRK2 were calculated using AutoDock Vina and visualized with PyMOL [25, 26]. The three-dimensional structures of human GRK5 (PDB 4TND) and GRK2 (PDB 5UUU) were downloaded from RCSB Protein Data Bank (PDB) and prepared using AutoDock Tools by removing all water and co-crystallized ligands, addition of polar hydrogens and assigning Kollman charges. Inhibitor isomeric simplified molecular input line entry system (SMILES) was obtained from PubChem and 3D coordinates generated using OpenBabel with the addition of hydrogens at pH 7.3 and partial charges calculated with a Gasteiger model.
The half maximal inhibitory concentration (IC50) of GRK5-IN-2 was determined using the ADP-Glo Kinase Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, a twofold serial dilution of GRK5-IN-2 was carried out in DMSO, and inhibitors were subsequently diluted into assay buffer to the final required concentrations. Each inhibitor dilution was transferred into a white 96-shallow well plate. GRK5 protein (final concentration at 0.5 mg/mL), ATP (final concentration at 25 µM), and casein (final concentration at 20 mg/mL) as the substrate were added to each well. Reactions were incubated for 120 min at room temperature. Then, ADP-Glo™ Reagent was added to each well and incubated at room temperature for 40 min to stop the kinase reaction and deplete the unconsumed ATP, leaving only ADP and a very low background of ATP. Kinase Detection Reagent was added and incubated at room temperature for 60 min to convert ADP to ATP and to introduce luciferase and luciferin to detect ATP using a plate-reading luminometer.
Adipogenesis, fatty acid uptake, and lipogenesis
To assess the effects of GRK5-IN-2 on adipogenesis, WT 3T3-L1 preadipocytes were differentiated as described in the section above (Cell cultures), and concurrently treated without (DMSO vehicle) or with GRK5-IN-2 (20, 40, and 80 μM) for 7 days. Fatty acid uptake and incorporation into lipids as well as de novo lipogenesis were determined using [3H]-oleic acid and [14C]-acetic acid, respectively, following the procedure adapted from our previous study [20]. Day 3 differentiated WT 3T3-L1 cell cultures were pretreated without (DMSO vehicle) and with GRK5-IN-2 (40 μM) for 30 min, and then labeled with 0.5 μCi of [1,2-14C]-acetic acid (PerkinElmer, Waltham, MA, USA) or 5 μCi of [9,10-3H(N)]-oleic acid (PerkinElmer) plus 0.04 mM oleic acid (Sigma-Aldrich) conjugated with 0.01 mM fatty acid free-bovine serum albumin (BSA) of DMEM supplemented with 10% FBS, 1% P/S and 1 μg/ml of insulin for 0 (no radioisotopes), 30, 60 and 120 min. Following radiolabeling, cells were washed with ice-cold DPBS twice and lipid-extracted with hexane:isopropanol (3:2, vol:vol). Lipid classes from standards and cellular lipid extracts were separated by thin layer chromatography using Silica Gel plates and a solvent system containing hexane:diethyl ether:acetic acid (80:20:2, vol:vol:vol). Lipids were visualized by exposure to iodine vapor, and bands corresponding to TAG, free cholesterol (FC), cholesteryl ester (CE), and phospholipid (PL) were scraped and counted using a scintillation counter. After lipid extraction, cell residue was dissolved with 0.1 N of NaOH, and protein concentrations were measured using a Pierce™ BCA Protein Assay Kit for protein normalization of data.
Statistics
Data are presented as mean ± standard error of the mean (SEM). All data points reflect biological replicates. Binary comparisons are performed using two-tailed Student’s t test. Datasets comparing the effect of a single independent variable on more than two groups are assessed by one-way ANOVA followed by Dunnett’s correction. Datasets containing groups defined by two independent variables (genotype, time) are assessed by two-way ANOVA with Sidak’s correction. Prism 10 software (GraphPad) is used to perform statistical analyses (Statistical significance p < 0.05) and generate graphical representations of data.

留言 (0)