
記住我
The concept of gene therapy dates back to the 1960s, but its clinical application became a reality in the 1990s. A pivotal moment occurred in 1990 when Dr. W. French Anderson conducted the first approved gene therapy trial, treating a young patient with adenosine deaminase deficiency using retroviral vectors to introduce a functional ADA gene into the patient’s T-cells (1). This breakthrough illustrated the potential for gene therapy to treat genetic diseases at their root cause, laying the foundation for future therapeutic developments.
Although this early success sparked widespread enthusiasm, the field faced significant setbacks in the late 1990s. Most notably, the tragic death of Jesse Gelsinger during a clinical trial for ornithine transcarbamylase deficiency in 1999 raised serious safety concerns and led to increased regulatory scrutiny (2). Nevertheless, gene therapy rebounded through technological advancements in vector safety and gene transfer efficiency during the early 2000s.
Building upon early successes and challenges, gene therapy has since evolved, driven by advancements in vector safety and gene transfer efficiency, positioning it as a cornerstone in modern oncology. Gene therapy is revolutionizing the field of oncology by offering precise, targeted treatments aimed at the underlying genetic causes of cancer. These therapies have emerged as a crucial component of modern cancer treatment, addressing the limitations of conventional treatment approaches like chemotherapy and radiation. Gene therapies enable personalized interventions, ranging from the correction of faulty genes to the enhancement of immune responses against tumors, thereby offering new avenues for treating both hematologic and solid malignancies (3, 4). In 2003, China approved Gendicine, the world’s first gene therapy for cancer, which delivers the p53 tumor suppressor gene to treat head and neck squamous cell carcinoma (5).
One of the most significant breakthroughs in gene therapy came with the advent of Chimeric Antigen Receptor T-cell (CAR-T) therapy, which modifies a patient’s T-cells to express receptors that specifically target cancer antigens. Although not approved as a first-line therapy, CAR T-cell therapy has transformed the treatment landscape for certain aggressive, relapsed, or refractory lymphomas and multiple myeloma.
Simultaneously, the discovery of the Clustered Regularly Interspaced Short Palindromic Repeats-Associated Protein 9 (CRISPR-Cas9) gene-editing system in 2012 represented a new era for gene therapy, allowing precise, targeted modifications to DNA sequences. CRISPR-Cas9 has since become a key tool in the development of gene therapies, enabling scientists to edit or disrupt specific genes with greater accuracy than previous technologies (6). By 2019, CRISPR-based therapies entered human clinical trials, further advancing the use of gene editing in cancer and other genetic disorders (7).
Today, gene therapy continues to advance with innovative approaches such as oncolytic virotherapy, which uses viruses to kill cancer cells selectively, and allogeneic CAR-T therapies, which provide “off-the-shelf” treatments using donor T-cells (8, 9). Together with advancements in gene-editing technologies like base editing and prime editing, these innovations hold the potential to overcome the limitations of current cancer treatments and expand the scope of gene therapy across oncology (10).
Decades of scientific breakthroughs and regulatory and clinical successes have positioned gene therapy as a cornerstone of modern cancer treatment (11). It offers hope for more effective, durable, and personalized cancer therapies in the future. Gene therapies have offered transformative solutions for patients with previously untreatable conditions. As Peter Marks, MD, PhD, Director of the Food and Drug Administration (FDA)‘s CBER, emphasized at the 2023 Cell & Gene Meeting on the Mesa: ‘We may not believe in miracles. But there are miraculous, and this is one.’ This highlights the regulatory focus on ensuring the safe development of gene therapies for small populations, with the goal of expanding access to broader groups in the future.
The landscape of gene therapy in oncology continues to evolve rapidly, driven by unprecedented advancements in precision medicine, gene-editing technologies, and innovative therapeutic strategies. With the increasing integration of immuno-oncology, gene therapies such as CAR-T, CRISPR-Cas9, and oncolytic virotherapy are expanding their potential in both hematologic and solid tumors. These therapies offer highly personalized, targeted treatments that not only address the genetic drivers of cancer but also aim to enhance immune responses, providing hope for more durable and effective solutions in cancer care. However, challenges such as tumor heterogeneity, immune evasion, and the complexities of the tumor microenvironment (TME) must be overcome to unlock the full potential of gene therapy. As the field continues to progress, the collaboration between gene therapy and immune modulation strategies represents one of the most promising paths forward, laying the foundation for the next generation of cancer therapies. This review will explore the current landscape of gene therapies in oncology, addressing both their groundbreaking potential and the challenges that must be surmounted to bring these therapies into mainstream clinical practice.
2 Types of gene therapy modalities in oncologyGene therapies in oncology offer several approaches to targeting cancer, each with distinct mechanisms of action. Recent advancements have expanded the scope and effectiveness of these therapies, improving patient outcomes by addressing critical challenges like precision, targeting, and delivery (Table 1).
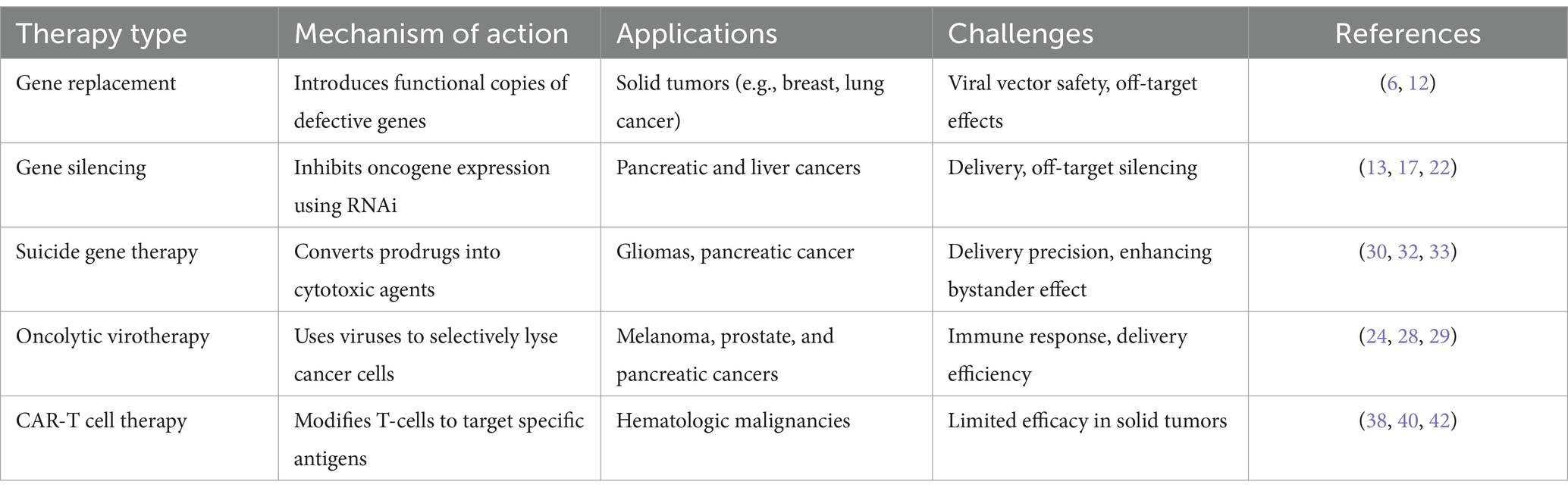
Table 1. Gene therapy modalities in oncology.
2.1 Gene replacement therapyGene replacement therapy introduces functional copies of defective or missing genes into cancer cells to restore normal cellular function. This therapy is particularly beneficial for cancers driven by specific genetic mutations, such as Tumor Protein 53 (TP53). Among the various viral delivery systems discussed later, Gendicine, an adenoviral vector (AdV) delivering the p53 gene, has been successfully employed in the treatment of head and neck squamous cell carcinoma. This achievement represents a notable milestone in advancing gene therapy for cancer (12). Recent advancements in adeno-associated viral (AAV) vectors have further improved the safety and targeting precision of gene replacement therapies, making them viable options for a wider range of oncological applications, including solid tumors like breast, lung, and colon cancers (6). These therapies utilize viral vectors such as AAV to deliver therapeutic genes directly to the tumor site, with the potential for durable responses and, in some cases, functional cures.
Moreover, the rise of CRISPR/Cas9 technology offers an additional layer of precision, enabling not only gene addition but also direct correction of mutations that drive tumorigenesis. Unlike traditional gene replacement methods that insert a functional gene copy, CRISPR-Cas9 allows for the in situ correction of genetic defects, including those involved in dominant-negative mutations, making it especially useful for cancers like colorectal carcinoma (4). However, while CRISPR presents an exciting opportunity for permanent correction of oncogenic mutations, challenges such as ethical concerns, off-target effects, and long-term safety still need to be addressed as discussed later.
2.2 Gene silencing therapyGene silencing therapy inhibits the expression of oncogenes that drive cancer progression, making it a critical approach in cancer treatment. RNA interference (RNAi) has emerged as a versatile and effective therapeutic strategy, with recent advancements in chemical modifications and delivery systems leading to the clinical approval of multiple siRNA-based drugs (13). These developments have improved the stability and efficacy of RNAi therapies while minimizing off-target effects (13, 14). RNAi, through small interfering RNAs (siRNA) or short hairpin RNAs (shRNA), target specific mRNAs for degradation, thereby preventing the production of dysfunctional proteins (15). For instance, targeting PLK1 via siRNA is being explored in clinical trials for pancreatic and liver cancers (16). Promising results from preclinical models of aggressive solid tumors further highlight the potential of RNAi-based therapies (17, 18). Additionally, gene silencing therapies using siRNA and miRNA have shown great potential in downregulating oncogenes that drive tumor progression (19, 20). siRNA targets specific mRNA sequences, preventing the production of proteins that fuel cancer growth. This approach has been applied in several cancers to silence genes involved in the aggressive types of cancer progression (2, 21). Furthermore, recent advances in the clinical translation of RNAi-based therapeutics have shown promise in downregulating oncogenes, offering a complementary approach to existing gene-editing technologies like CRISPR (22). miRNA therapy is also advancing in colon and lung cancers, playing a critical role in suppressing oncogenes and enhancing tumor suppression mechanisms (5, 23).
2.3 Oncolytic virotherapyOncolytic virotherapy uses genetically engineered viruses to infect and kill cancer cells selectively. The FDA-approved oncolytic virus Talimogene laherparepvec (Imlygic) has shown efficacy in treating melanoma (24). By infecting and lysing cancer cells, these viruses also stimulate an immune response, helping to attack remaining tumor cells (25). Researchers are now exploring oncolytic viruses in solid tumors like prostate and pancreatic cancers and enhancing the virus’s ability to penetrate tumors and amplify the immune response (26, 27). Recent studies, including Zou et al., (28) have demonstrated that combining oncolytic virotherapy with immune checkpoint inhibitors significantly amplifies the immune response against tumors, particularly in solid tumors where immune evasion mechanisms and the TME present significant challenges (28, 29).
2.4 Suicide gene therapySuicide gene therapy involves introducing genes into cancer cells that convert non-toxic prodrugs into cytotoxic agents, selectively inducing cell death within tumor tissues. One of the most well-established approaches utilizes the Herpes Simplex Virus Thymidine Kinase (HSV-TK) gene, which phosphorylates the antiviral drug ganciclovir, converting it into a toxic compound that kills dividing tumor cells (30). This strategy has shown promise in clinical trials, particularly for hard-to-treat cancers such as gliomas and pancreatic cancer (31, 32). In gliomas, the HSV-TK/ganciclovir system has been effective in reducing tumor volumes, primarily due to the bystander effect, where not only the genetically modified cells are killed, but also adjacent tumor cells. This effect occurs as cytotoxic metabolites diffuse from treated to neighboring cells, amplifying the therapeutic outcome (31, 32). This has led to promising results, especially when combined with conventional treatments like radiotherapy, which further sensitizes tumor cells to the cytotoxic effects of the prodrug (33).
Additionally, pancreatic cancer, a notoriously resistant solid tumor, has been a target of suicide gene therapy. Although early results have been encouraging, there is ongoing research to improve delivery methods and enhance the therapeutic effect. For instance, new delivery techniques using mesenchymal stem cells (MSCs) as carriers have been explored in preclinical studies (34, 35). These cells naturally home to tumor sites, improving the precision of suicide gene delivery and potentially enhancing therapeutic outcomes (36). MSCs can deliver the HSV-TK gene directly to the TME, ensuring localized activation of the prodrug and minimizing off-target effects However, challenges remain in increasing the efficacy of suicide gene therapy, especially in overcoming tumor resistance mechanisms, which limit the treatment’s success in aggressive cancers like pancreatic cancer. Additionally, while the bystander effect holds great potential for expanding the reach of the therapy beyond transduced cells, optimizing this effect remains a focus of ongoing research. Strategies to enhance the spread of cytotoxic metabolites and improve overall drug delivery are being actively investigated to increase the efficacy of HSV-TK-based therapies in broader tumor regions (37).
2.5 Autologous CAR-T cell therapySince its inception, CAR-T therapy has undergone significant evolution, as illustrated in Figure 1, CAR-T therapy emerging to improve efficacy and safety. The first-generation CAR-T cells featured a basic design with a single signaling domain (CD3ζ), primarily for T cell activation. In the second generation, costimulatory domains such as CD28 or 4-1BB were added, improving T cell expansion and persistence. The third generation further enhanced T cell functionality by combining multiple costimulatory domains. Fourth-generation CAR-T, also known as TRUCKs (T cells redirected for antigen-unrestricted cytokine-initiated killing), introduced cytokine signaling, such as Interleukin-12 (IL-12), to strengthen the immune response within the TME. The fifth generation incorporated even more sophisticated signaling pathways to better target tumors and counteract immunosuppression induced by the TME (38). Autologous CAR-T cell therapy involves collecting a patient’s own T cells, genetically engineering them to express a chimeric antigen receptor (CAR) targeting specific tumor antigens, and re-infusing them into the patient. This highly personalized immunotherapy has revolutionized the treatment of hematologic malignancies, particularly for relapsed or refractory B-cell malignancies, such as leukemia and lymphoma (38, 39).
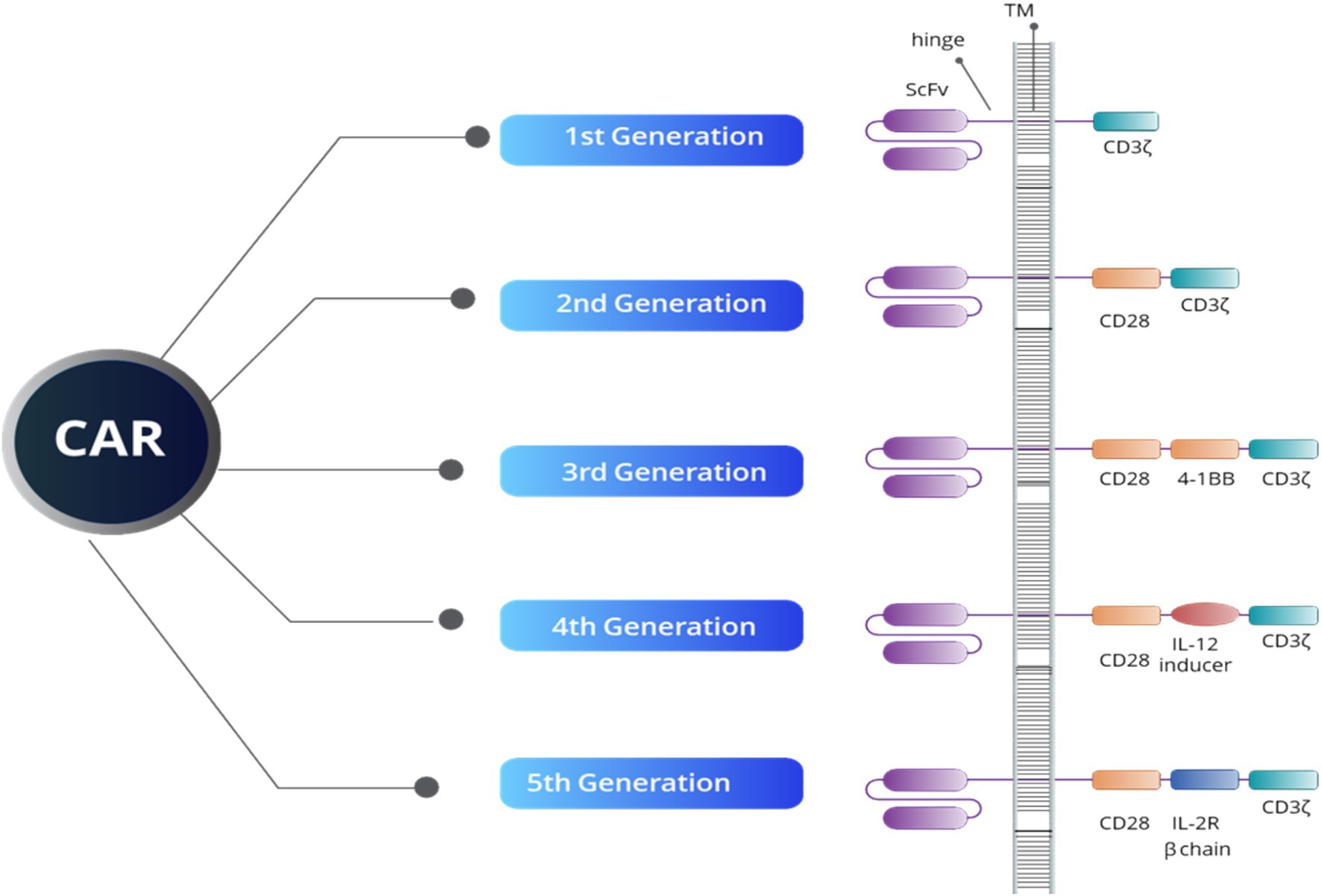
Figure 1. An overview of CAR structure reveals that all five generations of CAR constructs share four key domains: an extracellular domain that targets tumor-specific antigens (ScFV), a hinge region, a transmembrane domain (TM), and an intracellular domain. The intracellular domain’s structure defines both the generation of the CAR and its functional capacity. For example, the CD3ζ domain is crucial for initiating signal transduction pathways responsible for T-cell activation, proliferation, cytokine release, and cytotoxic activity. Additionally, the CD28 and 4-1BB domains serve as co-stimulatory signals, enhancing T-cell activation, longevity, and effectiveness. The IL-12 inducer domain facilitates cytokine production in the TME, while the IL-2R beta chain simulates IL-2 signaling, boosting CAR-T cell survival, proliferation, and persistence.
The FDA has approved several autologous CAR-T-cell therapies, including brexucabtagene autoleucel for adults with mantle cell lymphoma (MCL) and B-cell precursor acute lymphoblastic leukemia (ALL), tisagenlecleucel for pediatric and adult patients with relapsed or refractory ALL and large B-cell lymphoma, axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma and follicular lymphoma, and lisocabtagene maraleucel for relapsed or refractory large B-cell lymphoma. In multiple myeloma, Abecma (idecabtagene vicleucel) and Carvykti (ciltacabtagene autoleucel), both targeting B-cell maturation antigen (BCMA), have shown promise in treating relapsed or refractory cases, offering new options for patients who have exhausted conventional therapies (40–42). These therapies harness the patient’s immune system to attack and destroy cancer cells, but challenges remain, such as high manufacturing costs, logistical complexities, and limited efficacy in solid tumors (38, 39). Solid tumors, such as gliomas, present additional unique obstacles, further complicating the development of effective CAR-T therapies for these types of cancers due to their dense extracellular matrix and immunosuppressive microenvironment, which hinder the effective penetration and activity of CAR-T-cells. Recent studies suggest that optimizing CAR design could help overcome these obstacles. For instance, incorporating costimulatory domains such as 41BB and CD28 has enhanced CAR T-cell persistence and functionality in vitro. However, translating these improvements into in vivo settings remains a challenge (43, 44). A recent investigation into using B7-H3-specific CAR-T-cells for gliomas highlighted how CAR design can influence the recruitment and activation of immune cells, such as tumor-associated macrophages, within the brain TME (35). Such advancements suggest that further optimization of CAR-T cell therapies, mainly through engineering designs that modulate the immune microenvironment, may lead to improved outcomes in solid tumors like gliomas, where immune suppression and tumor heterogeneity continue to limit treatment efficacy (45).
2.6 Allogeneic CAR-T cell therapyAlthough autologous CAR-T therapy has demonstrated significant efficacy in treating certain cancers, it also presents challenges, including lengthy manufacturing times, high costs, and patient-specific variability. These issues are especially pronounced in patients with extensive prior treatments or compromised immune systems. To address these limitations, allogeneic CAR-T therapy, often referred to as “off-the-shelf” therapy, has emerged as a promising alternative (9). This approach uses donor-derived T-cells that are genetically engineered to express CARs and modified to avoid immune rejection by the recipient’s body (46). One of the key benefits of allogeneic CAR-T therapy is its ability to pre-manufacture and store CAR-T cells, making them readily available for use in multiple patients. This approach not only addresses scalability issues but also enhances the accessibility of these treatments compared to the more individualized autologous method. Furthermore, allogeneic CAR-T cells provide an option for patients with compromised immune systems or insufficient healthy T-cells—those for whom autologous treatments may not be feasible due to prior treatments or other health factors (47, 48). Additionally, allogeneic CAR-T cells offer faster production times, which is critical for patients with aggressive cancers who cannot afford to wait for the lengthy manufacturing process of autologous therapies (49).
In terms of cost, allogeneic CAR-T therapies enable bulk manufacturing, which significantly lowers production expenses and makes these treatments more affordable and accessible to a wider patient population. As such, allogeneic CAR-T therapy represents an important step forward in overcoming the limitations of autologous treatments and expanding access to life-saving cancer therapies (9, 50). Several allogeneic CAR-T therapies have entered clinical trials, demonstrating promising results in hematologic malignancies:
• UCART19: Developed by Cellectis, UCART19 is an allogeneic CAR-T therapy targeting CD19, a common antigen in B-cell malignancies such as ALL and B-cell lymphoma. Early-phase trials in patients with relapsed or refractory ALL have shown that UCART19 can induce remission, offering hope to patients who have exhausted other treatment options (51). The CALM clinical trial, initiated in 2016, evaluated UCART19 as an ‘off-the-shelf’ CAR T-cell product in adult patients with relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL). Final results, published in 2023, demonstrated a manageable safety profile and promising antileukemic activity, with 48% of treated patients achieving complete remission lasting an average of 7.4 months. Notably, patients who received alemtuzumab as part of the lymphodepletion regimen showed higher levels of UCART19 expansion and an improved disease response (52).
• ALLO-501 and ALLO-715: Developed by Allogene Therapeutics, ALLO-501 is an allogeneic CAR-T therapy targeting CD19, currently under investigation in patients with relapsed or refractory non-Hodgkin lymphoma (NHL). In parallel, ALLO-715 targets BCMA in patients with multiple myeloma. Both therapies have shown promising early results regarding safety and efficacy, suggesting that off-the-shelf CAR-T therapies could play a crucial role in the future of cancer immunotherapy (53). The ALPHA and ALPHA2 Phase 1 trials assessed ALLO-501 and its next-generation variant, ALLO-501A, in patients with relapsed or refractory NHL. Updated data presented in 2023 demonstrated that ALLO-501A has a manageable safety profile, with no dose-limiting toxicities or graft-versus-host disease (GvHD). Efficacy outcomes were comparable to those of autologous CAR T-cell therapies, with an overall response rate of 75% and a complete response rate of 53% across various histologies in CAR T-cell naïve patients.
• PBCAR0191: Precision BioSciences has developed PBCAR0191, an allogeneic CAR-T therapy targeting CD19. Preliminary clinical trials for patients with B-cell acute lymphoblastic leukemia (B-ALL) and NHL show encouraging responses with manageable safety profiles (53).
• Precigen’s UltraCAR-T® is utilizing an innovative system for a non-viral, multigene delivery process that allows for rapid, decentralized manufacturing, enabling same-day engineering and next-day infusion of T cells into patients. UltraCAR-T® cells are designed to express a (CAR, membrane-bound interleukin-15) (mbIL15) for enhanced persistence, and a safety kill switch, providing a robust framework to target both hematologic malignancies and solid tumors. For instance, PRGN-3005 targets MUC16 in ovarian cancer, while PRGN-3006 focuses on CD33 in AML.
• Additionally, early clinical trials with allogeneic CAR-T cells show promising efficacy in multiple myeloma and non-Hodgkin lymphoma (22). These advancements represent significant strides in improving the scalability, safety, and efficacy of CAR-T therapies, particularly in addressing the challenges of solid tumors and immune evasion.
Furthermore, switchable CAR-T cells have been developed to provide a controlled and safer therapeutic approach. This technology allows for the CAR-T cells to be turned on or off after administration, enabling more precise control over their activity and mitigating risks of uncontrolled proliferation and toxicity. This on/off mechanism generally involves administering an additional agent, such as an antibody or a small molecule, which acts as a “switch” to activate the CAR-T cells only when needed (54). Calibr’s CLBR001 CAR-T cells, in combination with their antibody switch SWI019, demonstrated promising phase 1 results with a high response rate and reduced duration of CRS and ICANS (RR). Similarly, AvenCell’s Universal Targeting platform uses a soluble targeting module to control CAR-T cell activity in cases of acute myeloid leukemia, reducing off-tumor effects associated with CD123-directed CARs (55). The advent of such technologies represents a significant stride toward safer, more effective CAR-T therapies, particularly in solid tumor environments, where immunosuppressive factors often induce CAR-T cell exhaustion, limiting efficacy.
Allogeneic CAR-T therapy represents a significant advancement in immunotherapy with great potential for future applications across multiple cancer types. Moreover, recent innovations in CAR-T engineering, including dual-targeting and switchable CARs, as well as the integration of checkpoint inhibitors to boost T-cell persistence in solid tumors, are demonstrating significant potential in preclinical and early-phase trials (40, 41, 56, 57). These innovative strategies are explored in greater detail in the Combination Therapies section.
2.7 Epigenetic modification in gene therapyEpigenetic modifications, which refer to changes in gene activity that do not alter the underlying DNA sequence, play a crucial role in cancer by influencing the activation of oncogenes, the silencing of tumor suppressor genes, and immune evasion mechanisms. Utilizing epigenetic tools, especially CRISPR/dCas9 systems, provides new avenues for cancer therapies that control gene expression without making permanent changes to the DNA. CRISPR/dCas9, a modified version of CRISPR-Cas9 that lacks DNA-cutting (endonuclease) activity, can bind to specific DNA regions to either increase gene expression (CRISPRa, for activation) or decrease gene expression (CRISPRi, for interference) (58–60). By attaching effector proteins that modulate epigenetic changes, such as DNA methylation or histone modification, CRISPR/dCas9 can precisely control oncogenes and tumor suppressors without altering the genome (Figure 2). This is particularly useful in oncology, where fine-tuning gene expression can enhance therapeutic outcomes (61). Furthermore, CRISPR/dCas9 can target multiple genes simultaneously, modulating key oncogenic pathways while activating tumor suppressors. This multi-target approach is effective in addressing tumor heterogeneity, resulting in more durable responses (61).
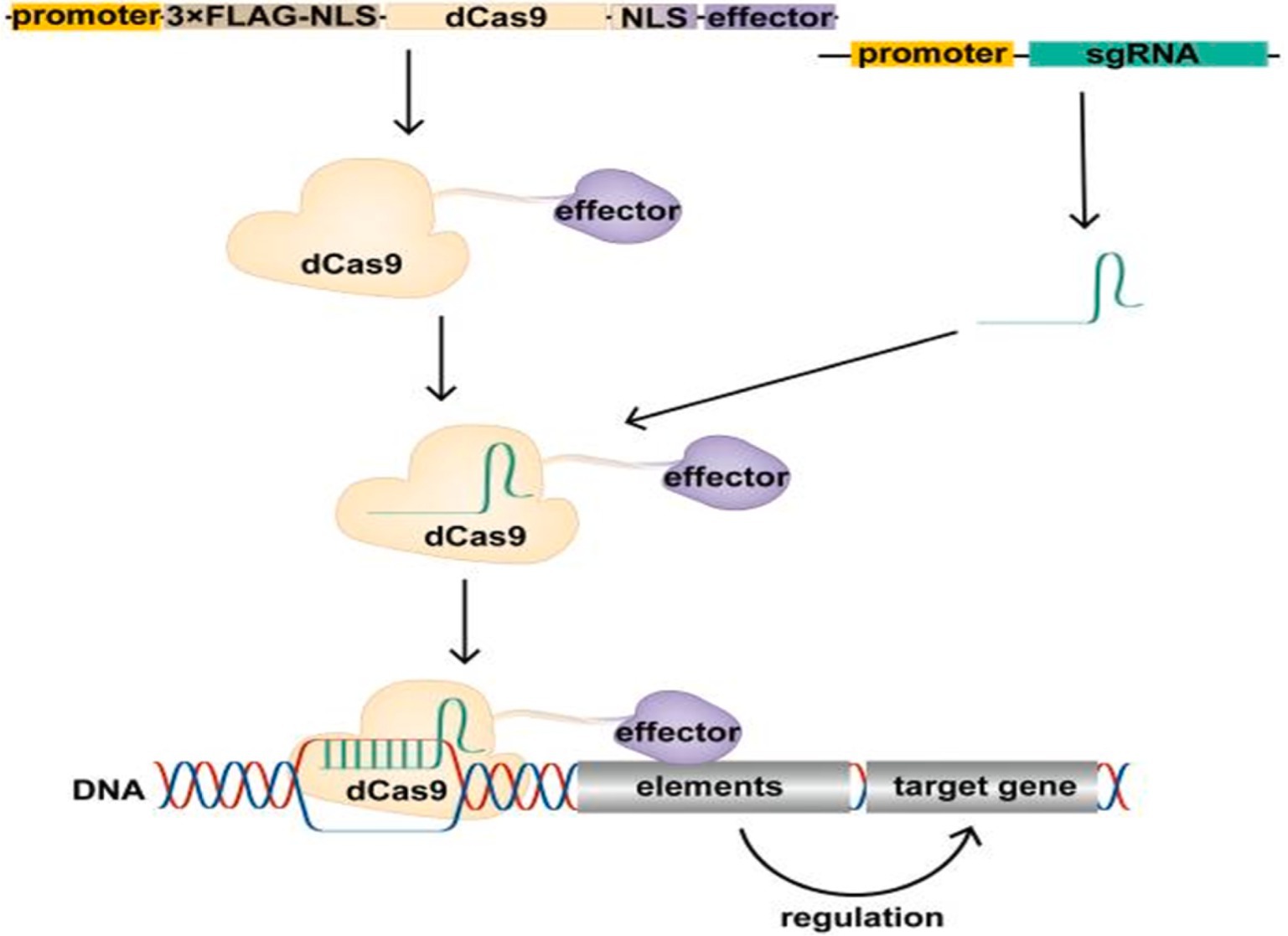
Figure 2. Schematic of the CRISPR/dCas9 regulatory target gene. The expression vector expresses the dCas9 fusion protein in cells, which binds the transcribed single guide RNA (sgRNA) to form the CRISPR/dCas9 regulatory tool, resulting in the recruitment of the effector domain to the promoter or enhancer region of the target gene under the guidance of sgRNA. Effector domains act on promoters or enhancers of target genes to modify these regions, regulating target gene expression. Above figure was adapted from Cai et al. (61).
Furthermore, CRISPR/dCas9 systems offer a way to modify immune-related genes in the TME, enhancing CAR-T cell infiltration and activity. CRISPRi can silence immunosuppressive genes like Programmed Death-Ligand 1 (PD-L1), while CRISPRa can boost immune-stimulatory genes, creating a more favorable environment for immune-based therapies (61). While epigenome editing is often considered safer than direct genome editing due to its non-alteration of the DNA sequence, it still necessitates thorough evaluation for both on- and off-target effects. This is because epigenome editing can influence multiple cellular pathways, potentially leading to unintended consequences. For instance, the use of epigenetic drugs has been associated with off-target effects and toxicity, highlighting the need for precision in these interventions (62). Moreover, unintended effects of epigenome editing on specific genomic regions may lead to toxic outcomes and influence human physiology, complicating its clinical application (63). Therefore, comprehensive testing is essential to ensure the safety and efficacy of epigenome editing technologies.
3 Delivery methods in gene therapyThe successful application of gene therapy depends heavily on the method used to deliver therapeutic genes to target cells. Delivery systems in gene therapy can be broadly categorized into viral, non-viral, physical, and live biotherapeutic products (LBPs). Each system has its strengths and challenges, and research is ongoing to optimize them for more effective and safe clinical applications.
3.1 Viral delivery systemsAs illustrated in Figure 3, viral vectors have been widely used in gene therapy due to their natural ability to infect cells and deliver genetic material efficiently. The most commonly employed viral vectors include:
• Adenoviral (AdV) vectors are highly effective in delivering genes to non-dividing cells; however, they can induce strong immune responses. To mitigate these effects and enhance their safety in clinical applications, engineering advancements are underway, including the development of helper-dependent adenoviral vectors that lack all viral coding sequences, thereby reducing immunogenicity and prolonging transgene expression (12, 64).
• Adeno-associated viral (AAV) vectors are highly efficient and generally considered safe due to their low immunogenicity. They are frequently used in both in vivo and ex vivo gene therapies, with applications ranging from hemophilia to cancer gene therapy (6, 65, 66).
• However, pre-existing immunity to AAV and limitations in payload capacity pose challenges. Strategies to overcome these obstacles include the development of novel AAV serotypes and engineered capsids to evade neutralizing antibodies, as well as the use of self-complementary AAV vectors to enhance transgene expression despite the limited packaging capacity (67).
• Lentiviral vectors, derived from HIV, offer stable integration into the host genome, making them suitable for long-term gene expression. These vectors are particularly favored in CAR-T cell therapies and stem cell modifications. Their ability to transduce both dividing and non-dividing cells, along with a relatively large packaging capacity, makes them versatile tools in gene therapy. Ongoing research focuses on improving their safety profiles by developing self-inactivating vectors and incorporating insulator elements to prevent insertional mutagenesis (38, 68).
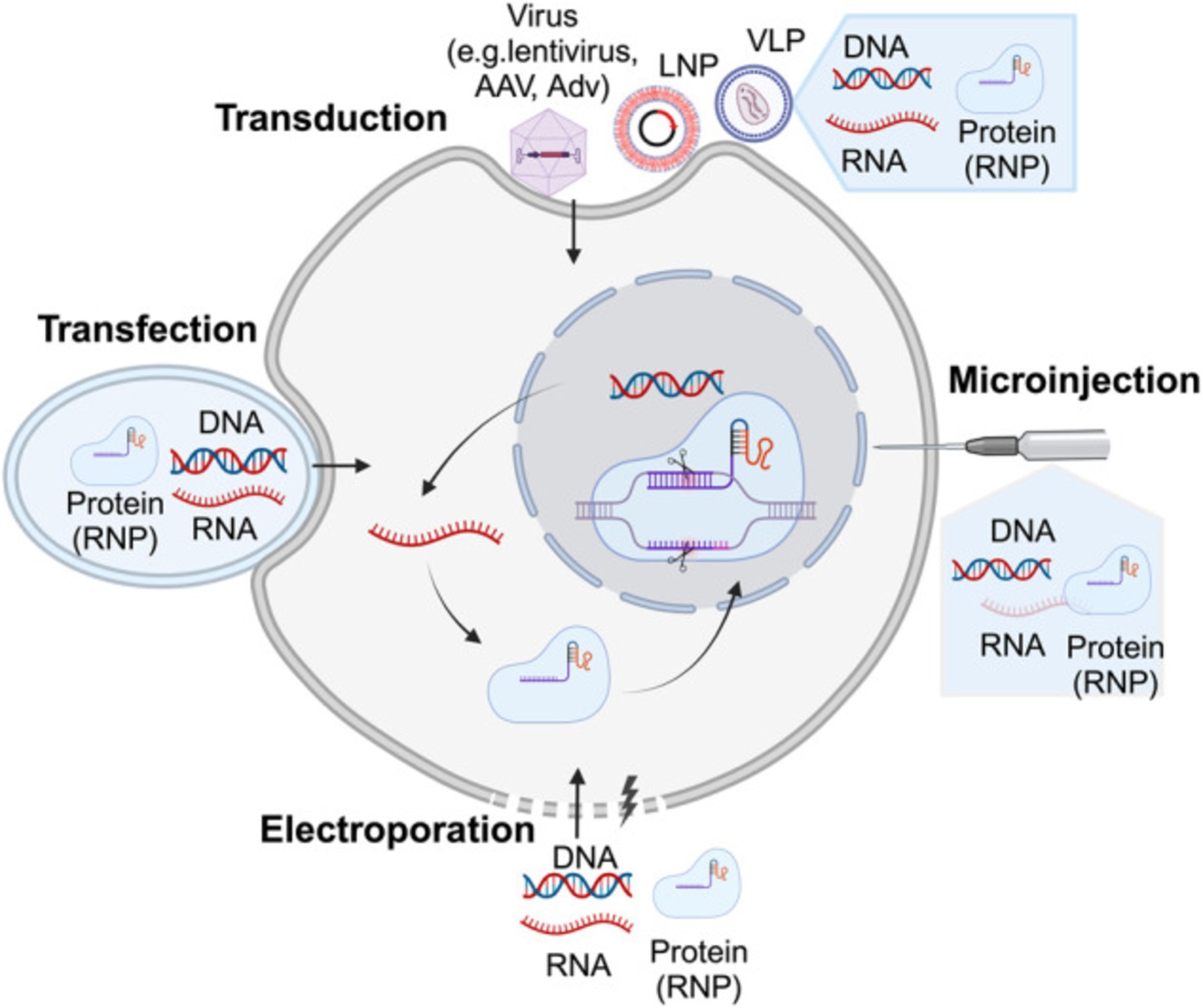
Figure 3. Illustrates the delivery strategies for precise genome-editing reagents. Precise genome-editing components encompass a variety of forms, including DNA, RNA, and protein complexes such as ribonucleoproteins (RNPs). DNA is commonly delivered through microinjection or electroporation of plasmids, as well as viral vectors such as lentivirus, AAV, and adenovirus (AdV). RNA can be introduced through microinjection or electroporation of RNPs, or via carriers like LNPs and virus-like particles (VLPs). Proteins, specifically RNPs, are typically delivered through microinjection or electroporation, or using carriers like LNPs and VLPs. The figure above was adapted from Zheng et al. (149).
3.2 Non-viral delivery systemsFigure 3 illustrates the non-viral delivery systems provide a safer alternative to viral vectors, especially in terms of immunogenicity and large-scale production, The diverse strategies for delivering genome-editing reagents include various forms such as DNA, RNA, and ribonucleoprotein complexes (RNPs). These components can be introduced into cells through techniques like microinjection, electroporation, or using viral and non-viral carriers such as lipid nanoparticles (LNPs) and virus-like particles (VLPs). Each delivery method is crucial for enhancing the precision and efficiency of genome editing, especially in therapeutic applications. These include:
• Lipid nanoparticles (LNPs): LNPs gained attention during the COVID-19 pandemic for mRNA vaccine delivery and are now being explored in gene therapy. LNPs encapsulate nucleic acids and other therapeutic agents, facilitating safe delivery with reduced immunogenicity. Their application in CRISPR-Cas9 delivery is particularly promising for cancer treatment (57).
• Polymeric nanoparticles: Synthetic polymers can deliver genes, small interfering RNA (siRNA), or other molecules into cells. Nethi et al. conclude that targeting ligand functionalization could be used to enhance the concentration of therapeutic Mesenchymal stem cells (MSC) constructs at the tumor tissue and to achieve improved antitumor response (69). Furthermore, recent studies focus on improving their targeting efficiency and reducing off-target effects, making them a valuable tool in solid tumor therapies (70).
• Exosome-mediated delivery: Exosomes, naturally occurring extracellular vesicles, offer a highly specific and less immunogenic method for delivering therapeutic payloads. As cell-derived vesicles, they possess inherent targeting capabilities, making them promising vehicles for gene delivery in cancer therapy. Recent studies have highlighted the potential of exosomes to transport a variety of genetic materials, including mRNA, siRNA, and CRISPR components, directly to cancer cells, even within the challenging TME (71, 72). Exosome-mediated delivery systems are particularly advantageous for their ability to bypass some of the common barriers faced by other delivery methods, such as immune activation and poor penetration into solid tumorsexosome-based delivery has been shown to enhance the stability and targeting specificity of therapeutic molecules. Preclinical studies have demonstrated that exosomes loaded with therapeutic RNA can successfully downregulate oncogenes and modulate immune responses in the TME, potentially improving treatment outcomes. Additionally, the low immunogenicity of exosomes reduces the risk of adverse immune responses, which is a significant concern with viral vector-based systems (73). Ongoing research exosome engineering, focusing on enhancing their loading capacity and targeting efficiency. Advanced techniques are being developed to modify the surface of exosomes with specific ligands, enabling precise delivery to cancer cells while sparing normal tissues. Clinical trials are underway to evaluate the safety and efficacy of exosome-based delivery systems, particularly in gene therapies targeting hard-to-treat cancers like pancreatic and glioblastoma (74).
3.3 Physical delivery methodsAs illustrated in Figure 3, physical methods use mechanical or physical means to deliver genetic material into cells. These techniques are particularly useful for ex vivo applications and are evolving to provide better precision and safety:
• Electroporation: This method uses an electric field to increase the permeability of the cell membrane, allowing genetic material to enter. It is especially effective in controlled ex vivo settings, where cells can be edited and reintroduced to the patient. Electroporation has been refined to improve the uptake of CRISPR components, reducing the risks of off-target effects associated with viral vectors (66, 75).
• Gene gun (biolistics): In this technique, high-pressure gas shoots particles coated with DNA or RNA into target cells. While its use is limited due to lower targeting precision, it is effective for tissues like skin and muscle.
• Ultrasound-mediated delivery: Ultrasound waves can temporarily increase cell membrane permeability, allowing genetic material to enter cells. This non-invasive method is under investigation for use in tissues such as the liver and muscles (76, 77).
• Hydrodynamic injection: In this technique, large volumes of solution containing therapeutic genes are injected at high pressure into the bloodstream. Though primarily used in preclinical studies, it has potential for liver-targeted gene therapies in humans (78).
3.4 Live biotherapeutic products (LBPs)LBPs represent an innovative delivery system that uses living microorganisms to deliver therapeutic genes. These are particularly effective in targeting the tumor TME:
• Bacterial delivery systems: Bacteria like Clostridium and Salmonella are engineered to proliferate in the hypoxic cores of tumors. These bacteria can deliver therapeutic genes or stimulate immune responses, improving treatment efficacy in cancer (59, 76).
• Probiotic-based gene delivery: Probiotics are genetically modified to deliver genes to specific tissues, such as the gut. This method offers a non-invasive delivery route and holds great promise for targeted therapies (79).
3.5 Combination of delivery methodsAs research continues, hybrid approaches that combine the efficiency of viral systems with the safety of non-viral systems are being developed. For example, hybrid LNP and viral vector systems aim to reduce immunogenicity while maintaining high delivery efficiency. These strategies are currently being explored in clinical trials, particularly in solid tumors, where overcoming barriers such as the dense extracellular matrix and immunosuppressive TME are significant hurdles (64, 65).
4 Challenges in gene therapy: CRISPR-Cas9 and advanced editingDelivering gene therapy effectively to cancer cells presents significant challenges. Each type of therapy requires precise targeting to ensure it reaches the appropriate cells without affecting healthy tissue. Additionally, overcoming physical and immunological barriers such as the tumor TME is essential for successful treatment, particularly in solid tumors. As discussed earlier, despite the promise of gene therapies, several challenges limit their widespread application, particularly in solid tumors:
• Targeting solid tumors: Solid tumors present a major challenge due to the dense extracellular matrix and immunosuppressive cells within the tumor TME, which inhibit the penetration and efficacy of gene therapies (56). Researchers are developing nanoparticle-based delivery systems and exosome-mediated therapies to improve tumor targeting and therapeutic delivery (71, 80). These approaches are being explored to enhance the precision of gene therapy in solid tumors, overcoming the dense extracellular matrix and immunosuppressive tumor TME (72, 76, 81). Furthermore, recent advancements in hybrid nanoparticle-viral delivery systems are optimizing gene therapy for solid tumors by enhancing delivery efficiency and minimizing immunogenicity (82).
• Off-target effects and safety concerns: Beyond these issues, allogeneic CAR-T therapy also shares several challenges with autologous CAR-T, particularly in the context of solid tumors. These include managing severe toxicities such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) (83). Additional hurdles like antigen escape, limited tumor infiltration, and the complex dynamics of the TME further complicate the therapeutic potential of CAR-T in solid tumors (84). However, recent advancements in CAR-T engineering, including refined gene-editing tools, are being actively developed to address these challenges and improve clinical outcomes in both hematologic malignancies and solid tumors (9, 85–87).
• Immune evasion: Dual-targeting CAR-T cells, engineered to recognize two antigens simultaneously, are being developed to reduce immune evasion by tumors. For instance, preclinical studies show that CAR-T cells targeting HER2 and IL13Rα2 antigens can effectively treat glioblastoma by overcoming tumor antigen heterogeneity and immune suppression within the TME. This strategy is also being explored in other solid tumors, such as NSCLC, to improve CAR-T cell infiltration and persistence (88).
• Scalability and cost: Many gene therapies, particularly CAR-T therapies, face challenges in scalability due to the labor-intensive and time-consuming process of harvesting and modifying patient cells. Innovations like automated CAR-T production and the development of allogeneic “off-the-shelf” therapies (e.g., ALLO-501, UCART19) are designed to reduce production time and make gene therapies more accessible to a broader population.
• Immune responses: Viral vectors, often used in gene therapy, can provoke immune reactions that compromise therapeutic effectiveness. As an alternative, non-viral delivery systems, such as LNPs, offer a promising solution. LNPs can be engineered to transport gene-editing tools, immune-stimulatory molecules, and chemotherapy agents directly to tumor sites, emerging as a safer option by reducing immune activation while ensuring efficient delivery. Clinical trials for solid tumors are currently exploring these approaches. However, RNA-based therapeutics, which typically require higher doses, present challenges such as toxicity and immunogenicity. In some cases, recipients have experienced pro-inflammatory responses, even at lower doses. Ongoing research is focused on optimizing LNP formulations to mitigate these side effects and enhance the safety of gene therapies (79).
• Durability of response: There are concerns regarding the durability of response in allogeneic CAR-T therapy. Early clinical trials have raised questions about the long-term persistence of donor CAR-T cells, as the recipient’s immune system may eventually clear these cells, potentially limiting their effectiveness (48). Efforts to enhance the longevity of allogeneic CAR-T cells are ongoing, with promising developments being made (89, 90).
• Secondary malignancies: The European Medicines Agency (EMA) has mandated that CAR-T therapies include warnings about the risk of secondary blood cancers, such as myelodysplastic syndromes and acute myeloid leukemia. This decision followed a safety review identifying cases of secondary T-cell cancers directly linked to CAR-T treatments (91, 92).
• Graft-Versus-Host Disease (GvHD): In allogeneic hematopoietic stem cell transplantation (allo-HSCT), GvHD remains a major barrier to long-term success. The pathophysiology involves donor immune cells attacking recipient tissues, leading to severe complications. Recent developments in prophylaxis and treatment aim to mitigate this risk, but GvHD continues to pose significant challenges in gene therapy applications (84, 93). To mitigate this, researchers are exploring methods such as TRAC and β2M gene knockouts, which aim to disrupt T-cell receptors and reduce the likelihood of immune responses from the host (85, 90).
• Another major challenge in gene therapy is the efficient delivery of therapeutic components, such as Cas9 and guide RNA, to target cells. Current research focuses on optimizing delivery methods, including viral vectors, nanoparticles, and electroporation, to enhance the safety and efficacy of CRISPR-based therapies (94, 95). Furthermore, CRISPR’s versatility in tackling complex genetic diseases, which often involve multiple gene interactions, enables simultaneous multi-gene targeting and the development of sophisticated genetic models that improve our understanding and treatment of these diseases (96).
As researchers continue to develop innovative strategies to overcome the inherent challenges of gene therapy, particularly in solid tumors, CRISPR technology has emerged as a transformative tool to address many of these obstacles. From improving precision and delivery methods to reducing off-target effects, CRISPR-based therapies offer significant advancements that mitigate the risks traditionally associated with gene therapies. By enabling precise genome editing and enhancing the delivery of therapeutic components, CRISPR not only tackles issues like off-target effects and immune responses but also improves the scalability and accessibility of gene therapies, particularly in complex cases (97, 98).
While CAR-T cells have been successful in blood cancers, their efficacy in solid tumors is limited. CRISPR/dCas9 can be used to improve CAR-T function by enhancing co-stimulatory molecules through CRISPRa or reducing inhibitory receptors like Programmed Cell Death Protein (PD-1) using CRISPRi. Additionally, CRISPR/dCas9 can target TME elements that restrict CAR-T infiltration, improving treatment outcomes (61). Moreover, CRISPR’s versatility in gene modulation, including CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa), provides dynamic therapeutic options without permanent genomic alterations. This flexibility is crucial in addressing the evolving landscape of gene therapy, offering a robust solution to the challenges discussed earlier, while paving the way for more efficient, scalable, and safer treatments.
In cases requiring temporary gene modulation, CRISPR technologies, such as CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa), offer reversible control over gene expression without permanent alterations, which is particularly advantageous for dynamic therapeutic needs (94, 95). Additionally, CRISPR plays a crucial role in mitigating immune responses that can arise from gene therapy delivery systems by reducing the immunogenicity of therapeutic vectors through engineering strategies and less inflammatory vector designs (99). CRISPR’s potential to streamline therapeutic development is further enhanced by its ability to lower the cost and scalability of gene therapy, making treatments more accessible, particularly for rare genetic disorders (100). Furthermore, CRISPR enables the establishment of more transparent regulatory frameworks by demonstrating clear efficacy and safety profiles through rigorous testing protocols, paving the way for smoother clinical translation (101).
CRISPR-Cas9 technology has dramatically enhanced its precision and therapeutic potential, focusing on minimizing off-target effects, refining editing methods, and expanding clinical applications. These breakthroughs have been driven by innovations in high-fidelity Cas9 proteins, such as SpCas9-HF1 and eSpCas9, which significantly reduce off-target cleavage by improving DNA recognition. Furthermore, base and prime editing techniques have emerged, allowing for precise DNA modifications without inducing double-strand breaks, which is crucial for therapeutic applications (102–104).
In parallel, gene-editing technologies like CRISPR-Cas9 and TALENs (Transcription Activator-Like Effector Nucleases) are also being employed to knock out the T-cell receptor (TCR) in donor cells. This prevents the immune system from recognizing them as foreign, thus reducing the risk of GvHD. By making donor T-cells universal, they can be used in any patient without the need for personalization, dramatically cutting down on production time while leveraging the precise editing capabilities of CRISPR-Cas9 and its derivatives (105).
In recent years, gene-editing technologies have revolutionized biomedical research, offering unprecedented control over genetic material. Among these tools, CRISPR-Cas9 has emerged as the most versatile and efficient, enabling precise modifications to DNA with remarkable ease compared to earlier methods like zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). This breakthrough has opened new avenues in both research and therapeutic interventions, particularly in the field of oncology, where CRISPR is being explored for its potential to enhance cancer treatments. As advancements continue to refine its precision, the impact of CRISPR-Cas9 on clinical applications has grown exponentially (102).
CRISPR’s therapeutic applications have also progressed significantly. While early CRISPR therapies were primarily ex vivo, in vivo applications are now advancing. For example, Intellia Therapeutics’ NTLA-2002 uses CRISPR-Cas9 directly within the body to edit genes responsible for hereditary angioedema, a rare genetic disorder related to inflammatory pathways (106). Furthermore, clinical trials involving CRISPR therapies have made strides in treating genetic disorders like sickle cell disease (SCD) and beta-thalassemia. In these trials, CRISPR is used to edit hematopoietic stem cells, boosting fetal hemoglobin production, offering the potential for a one-time cure. Notably, Exa-cel, a CRISPR-Cas9-based therapy for SCD and beta-thalassemia, is nearing regulatory approval in the U.S., marking a milestone for gene editing in medicine (107). In the realm of oncology, CRISPR is transforming cancer treatment through the development of immunotherapies. For instance, Iovance Biotherapeutics is using TALEN-modified, PD-1-inactivated tumor-infiltrating lymphocytes in trials targeting solid tumors (103, 104). Building on these advancements, CRISPR’s application in oncology is particularly promising, with its ability to target specific genetic mutations driving cancer. CRISPR-Cas9 has the potential to revolutionize cancer treatment by enhancing immune cell therapies and disrupting oncogenic pathways. Recent clinical trials have demonstrated its potential in modifying genes like PD-1 in T-cells, boosting antitumor activity. Additionally, emerging tools like base editing and prime editing allow single-nucleotide modifications without causing double-strand breaks, reducing the risk of off-target effects. These techniques are being explored to target key cancer mutations in genes like KRAS and TP53, both of which play critical roles in complex cancers (65, 92).
Gene-editing technologies like CRISPR-Cas9 offer promising advances but come with safety concerns, including off-target effects that may activate oncogenes or disrupt essential genes, particularly in immune cells, potentially leading to adverse immune responses or secondary malignancies (92). Additionally, personalized therapies such as CAR-T cell treatment are costly and time-consuming due to the need for harvesting and modifying patient-specific cells, limiting scalability and accessibility. Building on previous efforts, researchers continue to explore allogeneic, ‘off-the-shelf’ therapies using donor cells. However, these approaches come with known challenges, including compatibility issues and the risk of GvHD (84).
The future of gene therapy lies in integrating advanced delivery methods with immuno-oncology. CRISPR-edited CAR-T cells, designed to target multiple antigens, are emerging as a solution to overcome tumor heterogeneity and immune evasion in solid tumors. These dual-targeting CAR-T therapies, combined with improved delivery systems, pave the way for more effective treatments that can penetrate the TME and deliver precise genetic interventions (103).
5 Combination therapiesCombining gene therapy with other treatment modalities—such as immune checkpoint inhibitors, chemotherapy, and radiation—has shown great promise in overcoming limitations of individual approaches. Notably, pairing gene therapies with immune checkpoint inhibitors, which block proteins like PD-1 and Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4) to restore immune activity, has significantly enhanced immune responses in cancers like melanoma and lung cancer (108, 109). This combined strategy addresses key barriers such as immune evasion and tumor heterogeneity, ultimately improving the efficacy of treatments in solid tumors (110). Additionally, combining gene therapy with chemotherapy has demonstrated a synergistic effect. Chemotherapeutic agents not only debulk tumors but also induce immunogenic cell death, thereby enhancing the immune system’s ability to recognize and destroy cancer cells (111). Similarly, radiation therapy contributes to immune activation by triggering the release of pro-inflammatory signals, which sensitizes tumors to immune attacks (112, 113). These multi-modal approaches create a powerful strategy to optimize gene therapies, particularly in resistant cancers, by targeting various components of tumor biology and the TME. As research continues, combination therapies represent one of the most promising advancements in personalized cancer care.
Combination therapies in CAR-T cell treatments aim to enhance efficacy and reduce toxicity. By integrating CAR-T cells with immune checkpoint inhibitors, chemotherapy, or oncolytic viruses, these strategies improve antitumor responses and address challenges like immune suppression and tumor resistance. Such approaches optimize therapeutic outcomes, making treatments more effective and safer for patients.
5.1 CAR-T therapy combined with immune checkpoint inhibitorsCAR-T cell therapy has shown remarkable success in treating hematologic malignancies, but its efficacy in solid tumors has been limited due to the TME’s immunosuppressive nature. Recent clinical trials have focused on combining CAR-T therapy with immune checkpoint inhibitors to enhance CAR-T cell persistence and functionality in solid tumors.
For instance, a Phase I clinical trial combining CAR-T cells with nivolumab (a PD-1 inhibitor) in patients with advanced non-small cell lung cancer (NSCLC) showed improved T-cell infiltration and persistence in the tumor (45). Similarly, pembrolizumab (another PD-1 inhibitor) combined with CAR-T cells in patients with refractory ovarian cancer led to enhanced tumor penetration and clinical responses (40). These studies highlight the potential of immune checkpoint inhibitors to reduce T-cell exhaustion, a common obstacle in solid tumors, thus improving the antitumor activity of CAR-T cells (61, 114, 115).
5.2 CRISPR-edited CAR-T cells for multi-targetingRecent advances in CRISPR-Cas9 gene editing have enabled the enhancement of CAR-T therapies through multi-targeting strategies. Tumor heterogeneity, where different cancer cells within the same tumor express different antigens, often limits the efficacy of single-target CAR-T therapies.
Preclinical studies of glioblastoma have demonstrated that CRISPR-edited CAR-T cells targeting both Human Epidermal Growth Factor Receptor 2 (HER2) and IL13Rα2 antigens can overcome immune evasion by significantly reducing tumor growth and improving tumor control (61). Ongoing clinical trials are exploring dual-targeting CRISPR-edited CAR-T cells for hard-to-treat cancers like ovarian and pancreatic cancers, which are typically resistant to single-target therapies (9). This multi-targeting approach addresses antigen escape and increases the likelihood of sustained tumor control (61).
5.3 CAR-T therapy with oncolytic virotherapyOncolytic virotherapy, which uses genetically engineered viruses to selectively infect and lyse cancer cells, has shown promise when combined with CAR-T cell therapy. Beyond direct tumor lysis, these viruses prime the TME to enhance CAR-T cell activity. In preclinical models of pancreatic cancer, Zou et al. (28) demonstrated that this combination improved T-cell infiltration and prolonged survival (28).
Ongoing clinical trials are exploring this approach in solid tumors like glioblastoma and melanoma, with early data indicating enhanced immune responses and improved CAR-T efficacy in tumors previously resistant to immunotherapy (45). Oncolytic viruses also stimulate the immune system by releasing tumor-specific antigens, promoting a broader anti-tumor response. An FDA-approved oncolytic herpesvirus (Imlygic) has shown efficacy in advanced melanoma by lysing tumor cells and boosting immune recognition of residual cancer. Current research is extending these strategies by combining oncolytic viruses with immune checkpoint inhibitors to further enhance immune responses against solid tumors.
5.4 CAR-T therapy with radiation and chemotherapyRadiation therapy is
留言 (0)