
記住我
Immune checkpoint inhibitors (ICIs) have revolutionized the treatment of solid tumors and are already integrated in the standard treatment of advanced non-small cell lung cancer (NSCLC) (Antonia et al., 2017). At present, their implementation into the clinic is most advanced for ICIs targeting CTLA-4, PD-1 and the PD-1 ligand 1 (PD-L1) (He and Xu, 2020; Shiravand et al., 2022). To increase their therapeutic effectiveness, ICIs are frequently combined with conventional chemotherapy and/or radiotherapy (RT) (Yap et al., 2021; Vafaei et al., 2022; Walsh et al., 2023). These treatments exert direct cytotoxic effects on tumor cells, but also induce inflammatory responses and even prime antigen-specific immune responses, particularly when combined with ICIs. However, since there is still a significant proportion of non-responders and also resistance development, novel therapeutics targeting other immune checkpoints or immunosuppressive pathways in the tumor microenvironment are constantly being developed and tested in clinical trials (Zhou and Yang, 2023). One of these targets is the ecto-5′-nucleotidase CD73, a membrane-bound enzyme of the purinergic signaling pathway. Pro-inflammatory extracellular adenosine triphosphate (ATP) is degraded via the ecto-apyrase CD39 into adenosine monophosphate (AMP). CD73 further converts extracellular AMP into adenosine, which is known to balance inflammatory processes in normal tissues and to promote immune escape in the tumor microenvironment (Yang et al., 2021; Han et al., 2022; Bach et al., 2023; Kaur and Dora, 2023; Kowash and Akbay, 2023; Stagg et al., 2023).
Besides their beneficial use in reactivating an anti-tumoral immune response, ICIs are also associated with a new class of alarming toxicities termed immune-related adverse events (irAEs), which include ICI pneumonitis (Naidoo et al., 2020; Nishimura et al., 2022). This is of clinical importance, as thoracic radiotherapy can trigger radiation pneumonitis in sensitive patients. Although data from clinical trials to date suggest that the combination of chemo-radiotherapy and ICIs only slightly increases toxicity to the irradiated normal tissue, potential interactions that drive pulmonary adverse events in individual patients are not well understood (Antonia et al., 2017). Herein, it is increasingly acknowledged that heterogeneity in the sensitivity of individual patients to inflammation-associated adverse effects induced by radiotherapy, immunotherapy or their combination(s) depends not only on drug concentration, RT dose and quality, and treatment schedule, but is additionally influenced by patient-specific molecular, health, and environmental factors (Chen et al., 2021; Yamaguchi et al., 2022).
The results of the phase III PACIFIC trial (NCT02125461) established the current standard of care for patients with unresectable stage III NSCLC: platinum-based concurrent chemoradiotherapy (cCRT) followed by consolidation treatment with the anti-PD-L1 monoclonal antibody Durvalumab for 1 year (Antonia et al., 2017; 2018). Building on these findings, newer trials (i.e., COAST and PACIFIC-9) are exploring potential improvements by combining cCRT with dual immune checkpoint inhibition targeting both, PD-L1 and CD73. These adaptations could enhance the anti-tumor immune response and address immune evasion mechanisms that might limit the efficacy of PD-L1 monotherapy (Herbst et al., 2022; Bendell et al., 2023). Yet, increasing the complexity of combined treatment concepts by adding CD73-targeted immunotherapy will add to the disturbance of pulmonary immune homeostasis and the potential occurrence of overlapping irAEs in NSCLC treatment and also further complicate adequate risk assessment, diagnosis, and therapy of pneumonitis.
In this review, we assess how therapeutic intervention in CD73/adenosine dynamics could synergize with radiation-induced immune activation in combined radio-immunotherapy concepts to drive irAEs in the lung. Here, we focus on interactions between radiotherapy and CD73/adenosine-targeting strategies in subacute pneumonitis that need to be distinguished from potential overlapping chronic adverse effects (e.g., pulmonary fibrosis), which we have already reviewed elsewhere (De Leve et al., 2019a; De Leve et al., 2019b). In addition, we compare the incidence of (radiation) pneumonitis reported for the PACIFIC regimen, with and without additive targeting of CD73, to assess how this alters the risk for irAEs in the clinical setting. Finally, we draw clinically relevant conclusions, stressing the necessity of an individualized irAE risk profile assessment in the planning of combined radio-immunotherapies targeting CD73 and further research on this topic.
2 Immune-regulatory functions of CD73 and the purinergic signaling complexPurine nucleotides and nucleosides are crucial components of intracellular metabolic processes and nucleic acid biomolecules. In addition, especially adenine-based purines also mediate extracellular purinergic signaling, an evolutionally conserved pathway with multifaceted roles in homeostasis and disease, including the regulation of immune responses (De Leve et al., 2019a). In this section, we introduce the purinergic signaling complex, describe how CD73 mediates the resolution of immune responses and how its immune-regulatory function is harnessed in cancer treatment.
2.1 The purinergic signaling pathwayThe entirety of molecules involved in the purinergic signaling system comprises various channels and transporters for purine release and re-uptake, membrane-bound enzymes for extracellular purine hydrolysis, and respective signaling receptors for purine nucleosides and nucleotides, which collectively enable a high degree of signaling complexity (Figure 1) (Giuliani et al., 2021).
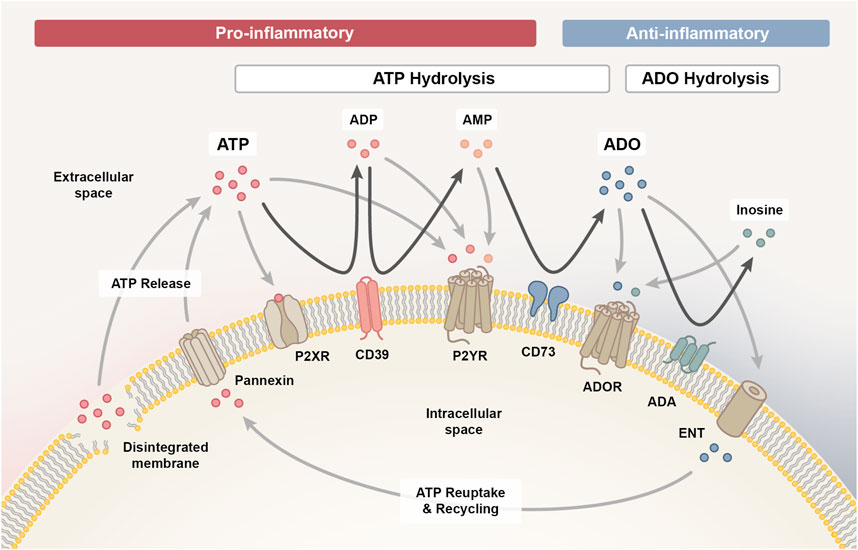
Figure 1. The purinergic signalling system. The purinergic signalling system comprises channels and transporters for purine release and re-uptake, membrane-bound enzymes for extracellular purine hydrolysis and respective P1, P2X and P2Y signalling receptors, which collectively coordinate the initiation and resolution of immune responses. Upon cellular damage, adenosine triphosphate (ATP) enters the extracellular space via pannexin channels or uncontrolled leakage. ATP signalling via P2X and P2Y receptors exerts predominantly pro-inflammatory effects, which facilitates the initiation of an inflammatory response required for the clearance of damaged cells. As the pro-inflammatory ATP pool is hydrolysed into adenosine diphosphate (ADP) and adenosine monophosphate (AMP) signalling intermediates via ectonucleotidase CD39 (ectonucleoside triphosphate diphosphohydrolase 1, ENTPD1) and further into adenosine (ADO) via CD73 (ecto-5′-nucleotidase, NT5E), the immune response transitions from the inflammatory towards the resolution and repair phase. AMP can also be generated through the non-canonical ENPP1 (ectonucleotide pyrophosphate phosphodiesterase 1) pathway (not shown), complementing the canonical CD39 pathway. Adenosine signalling via the P1 purinergic receptors A1, A2A, A2B and A3 primarily exerts anti-inflammatory effects on immune cells and is either reduced when adenosine is hydrolysed to inosine by membrane-bound adenosine deaminase (ADA) or re-entering the cell via concentrative or equilibrative nucleoside transporters (CNTs/ENTs) to be recycled to ATP. In addition, inosine can likewise interact with the A2A and A3 receptor to mediate anti-inflammatory responses.
Upon cell stress or damage, ATP can enter the extracellular space, either through uncontrolled leakage from disintegrated cell membranes or through controlled release via various means, such as pannexin 1 channels (Cekic and Linden, 2016). Extracellularly accumulating ATP functions as a danger signal and mediates paracrine or autocrine signaling via binding to ionotropic P2X or metabotropic P2Y receptors on the cell surface (De Leve et al., 2019b). P2X receptors are ligand-gated cation channels with seven different subtypes that function as homo- or heterotrimeric complexes specifically activated by ATP, while the eight recognized P2Y receptors are G protein-coupled receptors with differential specificity and affinity for ATP and ADP, as well as the respective uracil-based analogues, UTP and UDP (Kaur and Dora, 2023).
Signaling via P2X and P2Y receptors is terminated when extracellular ATP gets hydrolyzed into adenosine diphosphate (ADP), adenosine monophosphate (AMP) and adenosine signaling intermediates in a stepwise process mediated by membrane-bound ectonucleotidases. Initially, CD39 (ectonucleoside triphosphate diphosphohydrolase 1, ENTPD1) converts ATP into ADP and further into AMP (canonical pathway) (De Leve et al., 2019b). It is important to note that AMP can also be derived independent of CD39 via the CD38/ENPP1 axis (non-canonical pathway) (Ferretti et al., 2019). ENPP1/CD203a/PC-1 (ectonucleotide pyrophosphate phosphodiesterase 1) converts various substrates, including NAD+ (nicotinamide adenine dinucleotide), adenosine diphosphate ribose (ADPR), cGAMP and ATP, further contributing to the accumulation of AMP in the extracellular space (Ruiz-Fernandez de Cordoba et al., 2023). CD73 (ecto-5′-nucleotidase, NT5E) is the ectoenzyme ultimately hydrolyzing the remaining 5′ phosphate group from AMP to generate adenosine as the final product. Albeit, adenosine may also be directly released into the extracellular space from stressed or damaged cells (De Leve et al., 2019b).
Extracellular adenosine activates P1 purinergic receptors, a group of G protein-coupled receptors comprising four subtypes (A1, A2A, A2B and A3), that contrarily regulate adenylyl cyclase (AC). While A2A and A2B receptors are coupled to Gs proteins that activate AC to increase the production of intracellular cyclic AMP (cAMP), A1 and A3 receptors are coupled to Gi/o proteins that inhibit AC to reduce cAMP levels (Kaur and Dora, 2023). The purinergic receptor subtypes also differ in the signaling pathways they initiate within target cells and in adenosine affinity (A1 > A2A > A3 >> A2B), allowing adenosine to modulate both, routine and stress-induced cellular responses. While physiological adenosine levels are sufficient to activate A1, A2A and A3 receptors, activation of the A2B subtype requires tenfold higher adenosine concentrations, which only prevail in pathological or stressed conditions (De Leve et al., 2019b).
Signaling through P1 receptors recedes when extracellular adenosine is re-entering the cell via concentrative or equilibrative nucleoside transporters (CNTs/ENTs) to be recycled to AMP by intracellular adenosine kinase (ADK) (De Leve et al., 2019b). Alternatively, adenosine can also be deaminated by membrane-bound adenosine deaminase (ADA) to inosine, which likewise functions as an agonist for A2A and A3 receptors, albeit with lower affinity than adenosine and an extracellular-signal regulated kinases (ERK)1/2 downstream signaling bias (Gomez and Sitkovsky, 2003; Welihinda et al., 2016).
2.2 Purinergic regulation of immune responsesIn addition to its actions on cellular responses such as cell proliferation, differentiation, migration, apoptosis, neurotransmission, secretion, vasodilation and platelet aggregation, purinergic signaling is also crucial for the temporal coordination of immune responses. Most immune cells are equipped with components of the purinergic signaling system and their behavior is accordingly shaped by the cell-specific expression profile of purinergic receptors and ectoenzymes, as well as the local concentrations and ratio of ATP and adenosine. In this context, extracellular ATP primarily exerts pro-inflammatory effects, while adenosine has mostly anti-inflammatory or immunosuppressive properties (Cekic and Linden, 2016; Huang et al., 2021; Kaur and Dora, 2023).
2.2.1 Acute phaseIn the acute phase after tissue injury, the rapid accumulation of ATP released from stressed and damaged cells into the extracellular space facilitates the recruitment and activation of various immune cell types to initiate a robust immune response (Cekic and Linden, 2016). In particular, extracellular ATP functions as a chemoattractant for phagocytes (monocytes, macrophages, DCs and neutrophils) and stimulates immune cell activation. Signaling through P2X and P2Y receptors, which are highly expressed on monocytes and macrophages, stimulates activation of the inflammasome. Further, extracellular ATP promotes effector T cell receptor (TCR) signaling, while regulatory T cell (Treg) functionality and survival are diminished (Cekic and Linden, 2016).
2.2.2 Subacute phaseIn the subacute phase, the enhanced activity of CD39 and CD73 ectonucleotidases increases the local concentration of adenosine in order to limit the inflammatory response. Activated immune cells also upregulate the Gs-coupled P1 receptor subtypes A2A and A2B, which increases their sensitivity to extracellular adenosine. A2A receptors are expressed on the majority of immune cells (T cells, B cells, monocytes, macrophages, dendritic cells (DCs), natural killer (NK) cells, mast cells, eosinophils and platelets). In contrast, A2B receptors, which require higher adenosine concentrations for their activation, are primarily expressed on the surface of macrophages and DCs, and to a lower extent also on lymphocytes and platelets (Cekic and Linden, 2016). Adenosine signaling through A2A and A2B receptors activates AC to increase intracellular cAMP levels, which stimulate the production of anti-inflammatory cytokines (e.g., IL-10), while pro-inflammatory cytokines [e.g., tumor necrosis factor alpha (TNFα) or IL-1β] are downregulated (Wang et al., 2024). Further, adenosine inhibits phagocyte chemotaxis and activation, as well as antigen-presentation. Adenosine signaling limits effector T cell responses directly by suppressing cytokine secretion, T cell infiltration and de novo activation, but also by promoting exhaustion of already activated effector T cells (Wang et al., 2024). While adenosine restrains pro-inflammatory immune cell activities, it likewise fosters the expansion and polarization of tolerance-associated immune cell types, such as Tregs, M2 macrophages and myeloid-derived suppressor cells (MDSCs) (De Leve et al., 2019a). Besides its catalytic activity, CD73 on endothelial cells can also function physically as an adhesion receptor and is thus involved in orchestrating leukocyte trafficking in response to chemotactic stimuli and regulating vascular permeability (Eltzschig et al., 2004; Thompson et al., 2004; Salmi and Jalkanen, 2005; Linden, 2006; Eckle et al., 2007; Ålgars et al., 2011). Eventually, deamination of the accumulating adenosine pool by ADA generates inosine, which has a longer half-life than adenosine and can amplify and prolong its anti-inflammatory effects (Welihinda et al., 2016).
2.2.3 Chronic phaseAs the inflammatory response progresses further towards the chronic phase, the ATP/adenosine ratio continues to decline and mediates the initiation of wound healing processes. Yet, a prolonged increase in extracellular adenosine levels and hence persistent A2B receptor signaling at this stage can escalate to fibrotic tissue remodeling, mainly via upregulated secretion of IL-6 and vascular endothelial growth factor (VEGF), alternatively polarized macrophages and Th17 cells (Wirsdörfer et al., 2013; 2019; De Leve et al., 2017).
2.3 Pharmacologic targeting of CD73 to boost anti-tumor immunityCD73 and other components of the purinergic system are aberrantly expressed in numerous cancers and are considered as important regulators of an adverse tumor microenvironment. For example, chronically inflamed and hypoxic tumor microenvironments are associated with upregulated CD73 expression as well as deregulated levels of ATP and adenosine (Vaupel and Mayer, 2016; Chambers and Matosevic, 2019). In this regard, CD39 and CD73 play key roles in generating adenosine-enriched immunosuppressed and pro-angiogenic environments that support cancer development and malignant tumor (cell) behavior (Antonioli et al., 2016; Pacheco and Schenk, 2021). As adenosine-mediated immunosuppression constitutes one of many mechanisms utilized by tumors to escape from neoantigen-induced anti-tumor immune responses, overexpression of CD39, CD73 and A2A receptors in the tumor microenvironment further correlates with poor survival and therapy response. High intrinsic CD73/adenosine signaling in the tumor microenvironment may also restrain radiotherapy-induced antitumor immune responses and thereby limit the efficacy of combined radio-immunotherapies (Vaupel and Multhoff, 2016; Wennerberg et al., 2017). Consequently, adenosine receptor antagonism or pharmacologic inhibition of ectonucleotidase activity to restrain ATP hydrolysis and downstream generation of adenosine are promising strategies to improve the treatment of various solid tumors, including NSCLC. Furthermore, these factors are also used as prognostic biomarkers for various tumors (Wang et al., 2024).
CD73 is highly expressed in NSCLC and its expression correlates with and is regulated by common oncogenic drivers like Kirsten rat sarcoma virus (KRAS), epidermal growth factor receptor (EGFR), mitogen-activated extracellular signal-regulated kinase (MEK) or anaplastic lymphoma kinase (ALK) (Han et al., 2022; Saigí et al., 2023). Investigations on the pro-tumorigenic role of CD73 demonstrate that CD73 impacts various cancer hallmarks, such as tumor cell cycle progression, invasiveness, epithelial-mesenchymal transition (EMT), angiogenesis, migration and metastasis (Kowash and Akbay, 2023). In its function as an immune checkpoint, CD73 generates adenosine that establishes an immunosuppressive tumor microenvironment, primarily by interfering with effector T cell, DC and NK cell expansion and/or functionality. At the same time, CD73-derived adenosine fosters M2-polarized macrophages, Tregs and MDSCs (Kowash and Akbay, 2023; Saigí et al., 2023). While preclinical models revealed favorable anti-tumor effects of pharmacologic CD73 inhibition, treatment strategies combining CD73 blockade with standard chemo-radiotherapy or other immune checkpoint inhibitors warrant further investigation.
Various CD73-targeting pharmaceutics have proceeded to clinical testing, including the anti-CD73 monoclonal antibody (mAB) Oleclumab (MEDI9447), which selectively binds CD73 to inhibit its catalytic activity by steric blocking and dimer crosslinking and also increases CD73 internalization. The first in-human study of Oleclumab was a phase I clinical trial (NCT02503774) running from 2015 to 2023 that aimed to evaluate its efficacy and safety alone or in combination with Durvalumab (anti-PD-L1 mAB) in patients with advanced solid tumors (Bendell et al., 2023). At present, targeting of CD73 with Oleclumab is considered for combined radio-chemo-immunotherapy concepts to improve the survival of stage III unresectable NSCLC patients (COAST and PACIFIC-9 clinical trials) (Herbst et al., 2022; Barlesi et al., 2024).
3 Crossed immunomodulatory paths: How CD73-directed immunotherapy could exacerbate radiation pneumonitisPharmacologic targeting of immune checkpoints like CD73 has substantial therapeutic potential in cancer treatment, but in addition to a (re-)activation of the anti-tumoral immune response, ICIs can also trigger a specific class of toxicities, irAEs, which include ICI pneumonitis (Guberina et al., 2023). It is evident that ICIs are particularly effective when combined with conventional immune-activating therapies, such as radiotherapy. The therapeutic effect of radiotherapy is based on the complex local damage that ionizing radiation exerts to cellular macromolecules, particularly to the DNA, which commonly results in cell growth arrest or death. Yet, the discovery that ionizing radiation has also systemic effects and can boost anti-tumor immunity, introduced a paradigm shift and provides rationales for combining radio- and immunotherapies to achieve a synergistic treatment effect: These effects include, on the one hand, activation of local and systemic immune response mechanisms via classic damage-signaling cascades and immunogenic cell death [e.g., recognition of nuclear and mitochondrial DNA fragments via cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING), recognition of damage-associated molecular patterns (DAMPs) by, e.g., TOLL-like receptors (TLRs), immune-mediators from the senescence-associated secretory phenotype (SASPs) and inflammasome activation] and, on the other hand, enhancement of tumor antigen presentation (e.g., enhancing the exposure of immunogenic mutations frequently contained in DNA damage repair genes and increasing the release of tumor antigens upon cell death induction) (Wirsdörfer and Jendrossek, 2017; Lhuillier et al., 2019; Wirsdörfer et al., 2019; Guberina et al., 2023). Importantly, radiotherapy also mediates an increased cell surface expression of various immune checkpoints, including CD73, which represents an additional rationale for combined radio-immunotherapy (Mohamed et al., 2023).
Similar to ICIs, radiotherapy as such can thus also initiate irAEs. This observation is of particular clinical importance for thoracic radiotherapy, as exposure of the intrinsically radiosensitive normal lung tissue to ionizing radiation can trigger radiation pneumonitis, a late-occurring and potentially life-threatening immune-related complication of radiotherapy. The diagnosis of radiation pneumonitis is based on an increased density of the lung parenchyma in CT monitoring in conjunction with a recent history of thorax radiotherapy and the exclusion of other causes. Classic pneumonitis symptoms like cough, dyspnea, low-grade fever and chest pain commonly occur within 3–12 weeks after radiation exposure, but are not necessarily present. As these symptoms are non-specific and occurring with a delay, the clinical distinction between radiation pneumonitis and similarly presenting lung diseases such as ICI pneumonitis or pneumonia is challenging and contributes to the fact that radiation pneumonitis may not be recognized immediately in follow-up care (Bledsoe et al., 2017; Giuranno et al., 2019; Rahi et al., 2021; Guberina et al., 2023).
As the occurrence of radiation pneumonitis depends on patient-, tumor- and treatment-related factors, a wide range of clinical incidence rates for radiation pneumonitis is reported in the literature. For patients with stage III unresectable NSCLC treated with chemo-radiotherapy, the incidence of grade 3–5 radiation pneumonitis reported in a recent meta-analysis ranged from 3.62% to 7.85%, yet considerably higher incidence rates have been observed in vulnerable patient cohorts with underlying pulmonary dysfunction (Zhou Y. et al., 2020; Kuang et al., 2022). Although severe radiation pneumonitis is a rare complication of thoracic radiotherapy, mainly due to technical and physical innovations that have helped to enhance physical accuracy of dose delivery, it is nonetheless devastating for the prognosis of affected patients as only symptomatic treatment options are available. Thus, radiation pneumonitis remains a major dose-limiting factor.
With the addition of immuno-therapeutics to chemo-radiotherapy regimens, a novel, potentially overlapping risk factor for the development of pneumonitis has been introduced, which necessitates the re-evaluation of current risk assessments for each combinatorial treatment regimen. However, this type of investigation is still a largely neglected field of research in radiobiology.
Consolidation therapy with PD-1 and PD-L1 immune checkpoint inhibition has already been shown to increase the risk for radiation pneumonitis, as reviewed elsewhere (Chen et al., 2023). In the following paragraphs, we delineate the mechanisms driving radiation pneumonitis and elaborate how CD73-directed immunotherapy might interfere with disease pathogenesis or severity based on shared immunomodulatory pathways.
3.1 Radiation-induced immune response of the normal lung tissueRadiation-induced lung injury (RILI), which comprises subacute radiation pneumonitis (RP) and chronic radiation-induced lung fibrosis (RILF), is initiated by off-target radiation to non-immune and immune cells of the healthy lung tissue, causing an exaggerated immune response. The initial damage to cellular macromolecules, in particular the cellular DNA, can be induced directly, but up to 60% is induced indirectly via generation of reactive oxygen and nitrogen species (ROS/RNS), which can be even more detrimental, as they amplify, persist and spread to cells outside of the radiation field (Barcellos-Hoff et al., 2005; Azzam et al., 2012; Schipler and Iliakis, 2013; Giuranno et al., 2019). Based on cell-specific properties, the exposure to ionizing radiation leads to a cell type-specific damage load and stress response, with higher damage-loads usually triggering stronger immunogenic processes. At later stages, chronically alternating inflammation and tissue repair cycles without attaining actual resolution may cause progression to fibrotic tissue remodeling of the lungs, as nicely summarized by Jarzebska et al. (2021). The majority of the current knowledge on the mechanisms driving RILI has been gained in preclinical investigations in rodents and is described in the following paragraphs.
3.1.1 Acute phaseThe cellular response to the initial radiation damage, ranging from transient cell cycle arrest and recovery, over senescence and immunologically silent forms of cell death (e.g., apoptosis), to more immunogenic forms of cell death, is dependent on intrinsic cell properties (e.g., intrinsic radiosensitivity, DNA repair capacity and turnover kinetics), and decisive for the impact on the immune system (Denekamp and Rojas, 1989; Rosen et al., 1999; Golden et al., 2014; Bekker et al., 2022). Immune cells show a cell-type specific spectrum of radiosensitivity, but are in general considered to be highly susceptible to ionizing radiation. Hence, upon radiation exposure of the lungs, an acute but transient loss of various immune cells in the local tissue and also in the peripheral blood can be observed (Belka et al., 1999). This acute immune cell depletion is followed by a time-dependent immune cell recruitment, which establishes a sterile pro-inflammatory lung environment that facilitates the removal of injured cells to enable tissue restoration and the return to homeostasis (Belka et al., 1999; Heylmann et al., 2014; Wirsdörfer et al., 2014; Cytlak et al., 2022).
Radiation-induced immune activation is initially driven by classic tissue damage response pathways (mainly DAMP sensing, cGAS/STING-mediated DNA damage signaling and inflammasome activation) (Wirsdörfer et al., 2019): Shortly after damage induction (radiation), a spectrum of DAMPs is released from damaged cells into the extracellular space [e.g., ATP, high mobility group box chromosomal protein B1 (HMGB1), heat shock protein 70 (HSP70), uric acid, fragmented extracellular matrix molecules like low molecular weight hyaluronan (HA) and altered nucleic acids] or expressed on the cell surface [e.g., calreticulin (CRT)] (Land, 2015; Wirsdörfer and Jendrossek, 2017; Murao et al., 2021; Boerma et al., 2022). These damage signals are recognized by pattern recognition receptors (PRRs), such as TLR2 and TLR4, expressed on the surface of most non-immune cells [e.g., endothelial cells, alveolar epithelial cells (AECII) and fibroblasts] and also immune cells (e.g., monocytes, alveolar macrophages, neutrophils, DCs, NK cells and lymphocytes) (Jarzebska et al., 2021; Boerma et al., 2022). Damage sensing and PRR signaling initiate the sequential release of pro-inflammatory mediators, thereby fostering the time-dependent recruitment and activation of various innate and adaptive immune cells (Wirsdörfer et al., 2019).
Recruited immune cells respond with the production of further pro-inflammatory mediators. The cascade of inflammatory cytokines in the irradiated lung tissue can be further reinforced by activation of intracellular inflammasome protein complexes, for instance via binding of extracellular ATP to P2X7R purinoceptors. NOD-, LRR- and pyrine domain-containing protein 3 (NLRP3) inflammasome activation leads to the auto-catalytic cleavage of inactive pro-caspase-1 that in turn cleaves inactive precursors of the IL-1 family (IL-1β and IL-18), which bind to IL-1R-1, stimulating the production of further cytokines, like TNFα, IL-6 and also pro-IL-1β, that recruit and activate additional immune cells, establishing a robust sterile inflammation (Wirsdörfer and Jendrossek, 2017; Jarzebska et al., 2021). Inflammatory cytokines (TNFα, IL-6, type-I interferons) might also be induced upon sensing of altered cytosolic DNA species by cGAS, an intracellular PRR, which leads to phosphorylation of STING and the activation of nuclear factor kappa B (NF-κB) and interferon regulatory factor 3 (IRF3) (Yang et al., 2023).
In the course of the inflammatory response, dynamic immune cell composition changes and the transition from innate to adaptive immunity can be observed. As such, neutrophils and NK cells are attracted early by ROS and RNS species to initiate innate phagocytic and cytotoxic responses (Cytlak et al., 2022). Later, DAMPs function as a “find-me” signal, which is sensed by inflammatory monocytes and macrophages via their PRR to direct them to the site of injury and to encourage the removal of damaged cells. Further, the translocation of CRT to the cell surface of damaged cells undergoing immunogenic cell death in conjunction with HMGB1-TLR4 and ATP-P2XR7 damage signaling enables the maturation of DCs to prime adaptive immune cell responses (Elliott et al., 2009; Golden et al., 2014; Jarzebska et al., 2021; Cytlak et al., 2022).
3.1.2 Subacute phaseIn the subacute phase, the sterile inflammation in the lungs is reinforced via several mechanisms: As the inflammatory phase is marked by Th1 T cells, large amounts of pro-inflammatory cytokines like interferon gamma (IFNγ) are secreted, which, among other processes, promote the M1 polarization of macrophages that express inducible nitric oxide synthase (iNOS). Other pro-inflammatory mediators like TNFα and IL-1β activate iNOS to catalyze the production of large nitric oxide (NO) amounts, which cause secondary macromolecular damage that subsequently promotes further immune activation and amplification of the inflammatory response (Jarzebska et al., 2021; Yan et al., 2022). Other drivers of immune activation in the subacute phase are resident cells that acquired persistent sublethal radiation damage. As fully differentiated lung cells rarely divide, they are less prone to an immediate cell death upon exposure to ionizing radiation, but rather foster inflammation in the subacute phase when they eventually attempt cell division, which results in mitotic cell death due to previously acquired genome damages. Sublethal radiation damage can also induce permanent cell cycle arrest and the acquisition of a senescent phenotype, particularly in alveolar epithelial and endothelial cell populations. Senescent cells release a characteristic set of soluble mediators, termed the senescence-associated secretory phenotype (SASP), which contributes to the pro-inflammatory micromilieu (Citrin et al., 2013; Wunderlich et al., 2017; Hansel et al., 2020).
Continuous recruitment of immune cells is also promoted by radiation-induced damage to the alveolar endothelium via upregulation of adhesion molecules, like intracellular adhesion molecule 1 (ICAM-1) or vascular cell adhesion molecule 1 (VCAM-1), as well as structural changes to the endothelial glycocalyx, which facilitates the interaction with circulating leukocytes (Klein et al., 2016; Käsmann et al., 2020; Cytlak et al., 2022). The progressive loss of endothelial cells further results in vascular leakage and over time in oedema formation and chronic tissue hypoxia (Venkatesulu et al., 2018). Likewise, the loss of AECI and their reconstitution from AECII cells causes diminished production of alveolar surfactant, resulting in the loss of barrier function and increased alveolar surface tension in the long term. In addition, the dysfunction of AECII cells can cause deregulation of alveolar macrophages, which reside in close proximity to AECII cells for mutual regulation under homeostatic conditions (Bissonnette et al., 2020).
The above-mentioned amplification of the sterile inflammation and the immune-mediated secondary damage during the subacute phase requires counteracting anti-inflammatory mechanisms to keep the immune response under control. Herein, tolerance-associated immune cells like Tregs are suspected to counteract inflammatory mechanisms in the irradiated lungs by secreting various anti-inflammatory mediators, [e.g., transforming growth factor beta (TGFβ), IL-10 and IL-13], which suppress pro-inflammatory T cells and stimulate M2 polarization in macrophages (Wirsdörfer et al., 2014; Guo et al., 2020). In this context, also the CD73/adenosine axis can contribute to the resolution of inflammation and return to homeostasis (Figure 2). Early on in the acute radiation response phase of the lungs, extensive amounts of extracellular ATP are released, which functions as a DAMP and induces important pro-inflammatory responses, such as activation of the inflammasome and the subsequent release of inflammatory cytokines (Trautmann, 2009). In the subacute phase, this ATP pool is stepwise converted into adenosine by CD39 and CD73, which terminates P2X and P2Y signaling and enables the activation of P1 receptors. Own work using a murine whole thorax irradiation model revealed that adenosine levels in the lung tissue were significantly increased by week 16 after radiation exposure, and this was accompanied by an increased enzymatic activity of CD73 (Wirsdörfer et al., 2013). Research has demonstrated that adenosine affects a variety of immune cells, primarily mediating immunosuppression through negative feedback loop inhibition of activated immune cells and stimulation of anti-inflammatory subpopulations to stop further tissue damage (Sitkovsky et al., 2004). However, when pro-inflammatory cues continuously dominate and resolution fails, the inflammatory response of the lung tissue can exacerbate into subacute radiation pneumonitis.
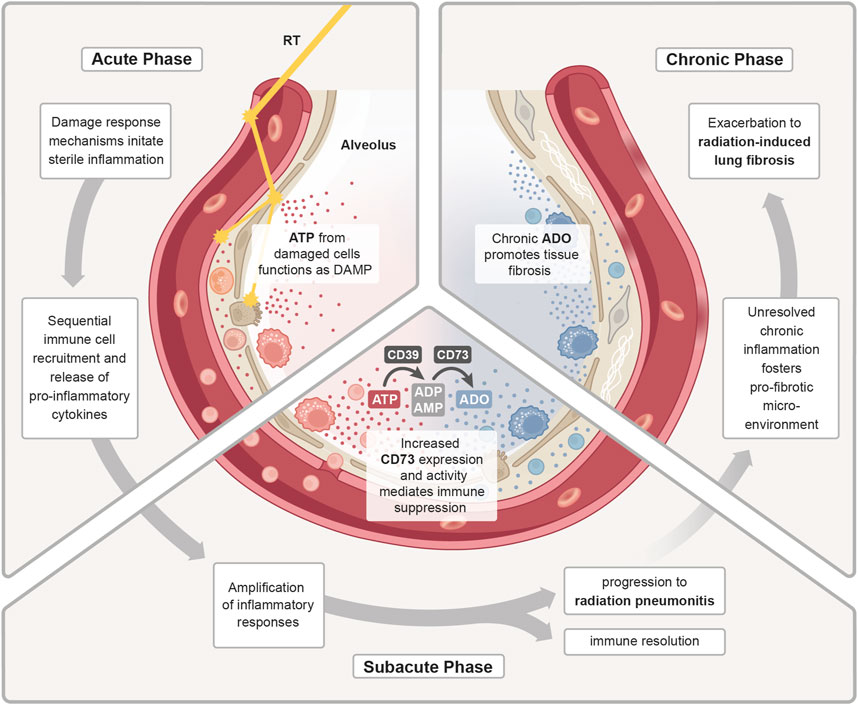
Figure 2. ATP/adenosine dynamics in radiation pneumonitis. Treatment with radiotherapy (RT) induces a damage response in the malignant and normal lung tissue. In the acute phase, release of adenosine triphosphate (ATP) from damaged cells functions as a DAMP to initiate a sterile inflammation. Subsequent immune cell recruitment and release of cytokines further fuels inflammation. During the sub-acute phase, the inflammatory response is further amplified and as a counteracting mechanism increased expression and activity of CD73 enhances the conversion of pro-inflammatory ATP into anti-inflammatory adenosine (ADO). The homeostatic regulation of inflammatory responses is needed to promote immune resolution and tissue regeneration. Uncontrolled, excessive pro-inflammatory responses can lead to radiation-induced pneumonitis. In the chronic phase, continuous cycles of secondary tissue damage and persistence of inflammatory drivers further changes the lung environment resulting in chronic inflammation and increase in profibrotic mediators (e.g., growth factors, TGFβ, hypoxia). Chronic ADO promotes this pro-fibrotic micromilieu which fosters fibroblast recruitment/activation and deposition of excessive extracellular matrix molecules resulting in radiation-induced lung fibrosis.
3.1.3 Chronic phaseWhen the ongoing inflammatory radiation response fails to reach true resolution, due to the persistence of inflammatory drivers and inflammation-induced secondary tissue damage, an imbalance of inflammation, counteracting immunosuppressive mechanisms and repair processes can cause fibrotic disease progression. Fibrogenesis is associated with accumulation of chronic adenosine and the pro-fibrotic key mediator TGFβ from multiple cellular sources and a switch from Th1/Th17 to Th2 T cell activities, which can stimulate the M2 polarization in macrophages via secretion of IL-4 and IL-13 (Yan et al., 2022). M2-polarized macrophages can in turn facilitate the activation of latent TGFβ pools. Ultimately, the pro-fibrotic microenvironment fosters an excessive secretion of extracellular matrix molecules (ECM) such as collagens from activated fibroblasts. Additionally, the chronic phase is also marked by an increase in pro-angiogenic factors, like vascular endothelial growth factor (VEGF) or hypoxia inducible factor 1 subunit alpha (HIF1α) (Bledsoe et al., 2017), which promote fibrosis and also stimulate futile angiogenesis.
3.1.4 Context-dependent function of CD73 in the inflammatory and fibrotic phases of radiation-induced lung injuryIt is assumed that the role of the CD73/adenosine axis in RILI varies significantly across the acute, sub-acute, and chronic stages, highlighting the context-dependent therapeutic potential of modulating CD73 activity. Both agonistic and antagonistic approaches may therefore offer benefits depending on the phase of disease progression (De Leve et al., 2019a; De Leve et al., 2019b).
In the acute phase of the radiation response, large amounts of extracellular ATP are released from damaged cells, acting as a chemotactic signal that recruits neutrophils, macrophages, and other immune cells to the injury site. These immune cells become activated to clear cellular debris. ATP also activates the NLRP3 inflammasome in macrophages via the P2X7 receptor, triggering the release of pro-inflammatory cytokines, particularly IL-1β and IL-18, which sustain the inflammatory response (Land, 2015; Wirsdörfer and Jendrossek, 2017; Murao et al., 2021; Boerma et al., 2022). Prolonged immune cell recruitment and high ATP levels amplify inflammation and contribute to bystander tissue damage. To counteract this, ATP is converted by CD39/CD73 ectoenzymes into immunosuppressive adenosine during the sub-acute phase, helping to limit excessive inflammation and prevent the development of chronic inflammation (Wirsdörfer et al., 2013). If unresolved, radiation-induced tissue damage, bystander effects, and leaky vasculature promote a persistent state of immune cell infiltration and activation. This chronic inflammation eventually polarizes macrophages from M1 to M2 phenotypes, contributing to a pro-fibrotic environment (Bledsoe et al., 2017; De Leve et al., 2017; Yan et al., 2022). In this context, own data show that loss of CD39 in a murine RILI model potentially fosters ATP-driven chronic inflammation resulting in bystander tissue damage and exacerbated radiation-induced lung fibrosis (Meyer et al., 2020). In the chronic phase, CD39/CD73-mediated ATP conversion likewise promotes pro-fibrotic signaling as sustained extracellular adenosine pools lead to chronic purinergic receptor stimulation leads to a transition from pro-inflammatory signaling to anti-inflammatory and tissue remodeling pathways (Wirsdörfer et al., 2013; De Leve et al., 2019a; De Leve et al., 2019b). Therapeutically, enhancing CD39/CD73-mediated ATP conversion to adenosine may be beneficial in the early phase to control inflammation, whereas antagonizing the purinergic system could help prevent or treat fibrosis in the chronic phase. Various pre-clinical studies have validated therapeutic strategies targeting purinergic signaling in lung injuries. These include modulating purinergic ectoenzymes and targeting purinergic receptors (Reutershan et al., 2007; Folkesson et al., 2012; Hoegl et al., 2015). Despite growing interest in targeting the purinergic system to treat lung injuries, studies specifically examining the role of this pathway in pneumopathies induced by radiation exposure are limited. Our own work with a murine whole-thorax irradiation model demonstrated a progressive increase in CD73 activity and adenosine levels post-irradiation. Notably, CD73-deficient mice showed reduced fibrosis, and pharmacological inhibition of CD73 or adenosine deaminase significantly mitigated fibrosis. This underscores the therapeutic potential of targeting adenosine signaling to alleviate radiation-induced lung damage (Wirsdörfer et al., 2013). Additionally, the adaptive transfer of CD73+ mesenchymal stem cells (MSCs) or their secretory products, such as extracellular vesicles (EVs), has shown promise in pre-clinical thorax irradiation models by reducing vascular damage, inflammation, and fibrosis in the lungs (Klein et al., 2016; Klein et al., 2017; Lei et al., 2021; Montay-Gruel et al., 2021; Shao et al., 2021; Hou et al., 2022; Li et al., 2022; Gupta et al., 2024). While CD73 is a required identification marker for MSCs (Dominici et al., 2006), the role of MSC-derived CD73 in RILI remains unclear. Moreover, the immunosuppressive potency of human MSC-EV preparations intended for clinical applications has not been directly attributed to the presence of CD73 (Bauer et al., 2023).
In the context of combined radio-immunotherapies for stage III unresectable NSCLC, CD73 is targeted with intent to sustain radiation-induced immune activation through extracellular ATP. However, the conversion of ATP to adenosine within the tumor microenvironment suppresses DC maturation and inhibits CD8+ cytotoxic T cells, thereby weakening antigen presentation and the anti-tumor immune response (Mastelic-Gavillet et al., 2019; Wennerberg et al., 2020; Lin et al., 2023). Furthermore, CD73 activity enhances tumor cell survival, migration, and invasion via adenosine-mediated activation of signaling pathways such as phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and mitogen-activated protein kinases (MAPK) (Ma et al., 2019; Zhan et al., 2024). As discussed earlier, the consequences of CD73 modulation in RILI will be highly context-dependent. In current clinical trials (e.g., COAST) administration of the CD73-targeting antibody Oleclumab begins 1–42 days after cCRT completion, thus coinciding with the acute and sub-acute post-irradiation phase. However, as immunotherapy is administered as consolidation treatment for up to 1 year, there is also potential for an overlap with the onset of chronic adverse effects, such as fibrosis (Herbst et al., 2022).
In the following section, we explore how therapeutic inhibition of CD73 activity might influence radiation-induced pneumonitis by modulating the immune response. These effects must be differentiated from the potential benefits of CD73 antagonism during chronic tissue remodeling. For comprehensive reviews of the role of CD73/adenosine in radiation-induced tissue fibrosis, refer to De Leve et al. (2019b), De Leve et al. (2019a).
3.2 Perspective: how therapeutic targeting of CD73 might interfere with immune regulation in radiation pneumonitisTherapeutic intervention in the purinergic system could exert multiple immunomodulatory effects in the irradiated lung. In the malignant tissue, where CD73 is already upregulated or induced by radiation, purinergic adenosine production leads to an immunosuppressive adenosine “halo” around the tumor that mediates immune escape as described above and in other recent reviews (Yang et al., 2021; Allard et al., 2023; Bach et al., 2023; Xia et al., 2023). So far, it remains to be determined if CD73-mediated immunomodulation plays a significant role in radiation pneumonitis. In the co-irradiated normal lung tissue, radiation induces a damage response and immune infiltration and subsequent inflammation, potentially exacerbating into radiation pneumonitis. While the acute response to ionizing radiation is dominated by extracellular ATP, the subacute and chronic phase are associated with a decline in the ATP/adenosine ratio (Volmer et al., 2006; Wirsdörfer et al., 2013; Jarzebska et al., 2021). This is a consequence of the inflammation-induced upregulation and increased activity of CD73 on diverse resident lung cells, as well as immune cells in the normal lung tissue, thus contributing to elevated adenosine levels, presumably to dampen overwhelming inflammatory responses and limit radiation pneumonitis as shown in a murine model of radiation-induced pneumopathy (Wirsdörfer et al., 2013). Depending on its extracellular concentration, adenosine will act on its receptors (A1, A2A, A2B and/or A3) to reduce the infiltration of inflammatory cells, dampen the release of pro-inflammatory cytokines, and promote tissue repair. Thus, controlled and transient CD73 activity will serve as a critical modulator that helps to mitigate lung damage and inflammation induced by radiotherapy. However, the above-mentioned acute protective effects are distinct from chronically increased CD73/adenosine signaling, which exerts adverse effects by promoting pathologic macrophage polarization, exaggerated matrix deposition chronic pulmonary fibrosis (De Leve et al., 2019b).
It is therefore important to take into account that a combined therapeutic intervention with thoracic irradiation and an anti-CD73 mAB might not only synergize in anti-tumor immunity and limit chronic toxicities (fibrosis), but also synergize in subacute normal tissue toxicities such as radiation pneumonitis to exert stronger adverse effects, if not compensated by other mechanisms (De Leve et al., 2019b). Therapeutic intervention in the CD73/adenosine signaling pathway is therefore a double-edged sword and should be planned with care.
In the following, we explore overlapping immunomodulatory effects of thoracic radiotherapy and CD73-directed immunotherapy on diverse cell types with relevance to lung injury/pneumonitis. As depicted in Figure 3, CD73/adenosine signaling on immune cells like neutrophils (Cronstein et al., 1985; Cronstein et al., 1990; Richter, 1992; Zalavary et al., 1994; Thiel and Chouker, 1995; Walker et al., 1997; Haskó and Cronstein, 2004; Linden, 2006; McColl et al., 2006; Eltzschig et al., 2008; Säve et al., 2011; Barletta et al., 2012; Petrovic-Djergovic et al., 2012), macrophages (Szabó et al., 1998; Németh et al., 2005; Kreckler et al., 2006; Ramanathan et al., 2007; Csóka et al., 2008; Chen et al., 2009; Buenestado et al., 2010; Ernens et al., 2010; Belikoff et al., 2011), MDSCs (Ryzhov et al., 2011; Sarkar et al., 2023), NK cells (Raskovalova et al., 2006; Young et al., 2018; Chambers and Matosevic, 2019), DCs (Addi et al., 2008; Novitskiy et al., 2008; Wilson et al., 2009; Yang et al., 2010; Silva-Vilches et al., 2018; Wennerberg et al., 2020), Tregs (Bopp et al., 2007; Deaglio et al., 2007; Dwyer et al., 2007; Zarek et al., 2008; Nakatsukasa et al., 2011; Ehrentraut et al., 2012; Kinsey et al., 2012; Ohta et al., 2012; Boveda-Ruiz et al., 2013; Schuler et al., 2014), T cells (Erdmann et al., 2005; Lappas et al., 2005; Csóka et al., 2008; Takedachi et al., 2008; Cekic et al., 2013; Leavy, 2013; Cekic and Linden, 2014; Kjaergaard et al., 2018; Leone et al., 2018; Vigano et al., 2019), B cells (Saze et al., 2013; Schena et al., 2013; Przybyla et al., 2018; Jeske et al., 2020) and lung resident non-immune cells like endothelial (Salmi and Jalkanen, 2005; Eckle et al., 2007; Umapathy et al., 2010) and epithelial cells (Lazarowski et al., 1992; Picher et al., 2003; Factor et al., 2007) is related to impaired migration, proliferation, antigen-presentation, pro-inflammatory cytokine and chemokine secretion, as well as impaired cytotoxicity and other anti-tumor responses and would therefore counteract radiation-induced pneumonitis. The therapeutic intervention with an anti-CD73 therapy has therefore the potential to reduce adenosine production and signaling and as a consequence limit the mentioned changes in the depicted cells. In the irradiated (damaged) normal tissue, pro-inflammatory responses are consequentially not dampened to a similar extent and, dependent on the severity of damage or immune response, can result in exaggerated inflammation and stronger radiation pneumonitis. Unfortunately, data on therapeutic targeting of CD73 in the context of acute and subacute radiation-induced lung toxicities is very limited. The available studies that investigate the role of CD73/adenosine targeting mostly show the therapeutic effects on tumor responses, thus we summarize here the major immunomodulatory actions of CD73/adenosine and its intervention and discuss a potential impact on toxic side effects in the lungs in the following paragraph.
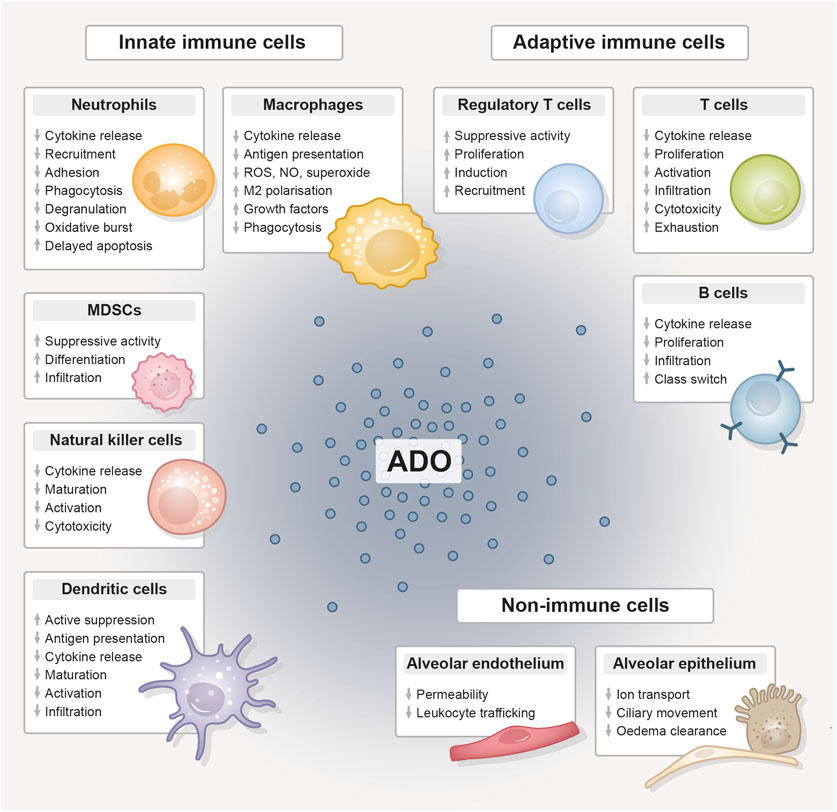
Figure 3. The immunomodulative function of extracellular adenosine. Compilation of the immunomodulative function of extracellular adenosine (ADO), generated from adenosine triphosphate (ATP) by the abundantly expressed CD39/CD73 ectoenzyme machinery, on innate and adaptive immune cell populations and lung-resident non-immune cells. Adenosine unfolds its anti-inflammatory effect primarily by stimulating tolerance-associated cell types, while the functionality and expansion of other immune cells is inhibited. ADO, adenosine; MDSC, myeloid-derived suppressor cell; ROS, reactive oxygen species; NO, nitric oxide.
For antigen-presenting cells like DCs, it was shown in a murine breast cancer model that an anti-CD73 therapy combined with radiotherapy restored conventional DC (cDC) activity and their infiltration into the tumor, enhanced the CD8+/Treg ratio and improved tumor control (Wennerberg et al., 2020). However, the positive reactivation or cancellation of suppression could have negative consequences in the irradiated normal tissue. The irradiated and inflamed lung microenvironment also influences the phenotype of lung DCs. Dependent on the micromilieu, DCs can be differentiated into TNF-DCs, IFN-DCs, (thymic stromal lymphopoietin) TSLP-DCs, IL-15-DCs, chemokine C-C-motif ligand 19 (CCL19)-DCs, or chemokine CXC-motif ligand 4 (CXCL4)-DCs by TNF-α, IFNγ, IL-15, IL-10, CCL19, and CXCL4, respectively (Liu et al., 2023). For instance, TSLP-DCs secrete high levels of type 2 cytokines and TNFα (Tormo and Gauchat, 2013). IFNγ-DCs increase the release of IL-12, which facilitates an efficient T-cell response (Gagliostro et al., 2016). CXCL4-DCs promote the generation of IFNγ and IL-4 as well as the growth of autologous CD4+ and CD8+ T cells (Silva-Cardoso et al., 2020; Gowhari Shabgah et al., 2021). Moreover, monocyte-derived inflammatory DCs (inf-DC, also called mo-DC) will also be present in the irradiated lung tissue, which derive from circulating Ly6Chigh monocytes that are considered to be the direct precursors of inf-DCs (Merad et al., 2013). These inf-DCs produce high amounts of TNFα and NO, thus also called “TNFα and iNOS producing” Tip-DC (Serbina et al., 2003; Cook and MacDonald, 2016). Thus, we speculate that, dependent on the individual lung milieu, anti-CD73 therapy may impact specific DC subsets and thereby inducing stronger pro-inflammatory cytokine and ROS production and enhancing the risk for radiation pneumonitis.
The same holds true for other innate immune cells like neutrophils and macrophages. A modulation of CD73 activity can alter macrophage functions by switching M1 and M2 phenotypes and also downregulate neutrophil activity (Haskó and Cronstein, 2004; Eltzschig et al., 2008; Yegutkin et al., 2011; Zanin et al., 2012; Ponce et al., 2016). As a consequence of targeted anti-CD73 therapy, both cell types could show a more pronounced pro-inflammatory phenotype and a series of cytokines, such as TNFα, IL-1β, IL-6, and TGFβ, could be released and enhance pneumonitis (Yarosz and Chang, 2018).
NK cells, important mediators of tumor-killing, have low levels of CD73 expression. Nevertheless, this expression is significant in tissues that have been invaded by tumors, indicating that NK cells may be able to inhibit the immune system by producing adenosine, if certain environmental conditions are met (Chatterjee et al., 2014). Adenosine generated by CD73 mainly inhibits NK cell activities via A2A (Raskovalova et al., 2006; Wang and Matosevic, 2018), thus leading to impaired maturation, activation, and cytotoxic potential of NK cells (Raskovalova et al., 2006; Beavis et al., 2013; Hatfield et al., 2015; Young et al., 2016; Chambers et al., 2018); in addition, NK cells in the tumor microenvironment undergo transcriptional reprogramming and upregulate IL-10 production (Neo et al., 2020). A study from Wang et al. showed in a murine model of radiation pneumonitis that radiation pneumonitis was accompanied by an accumulation of NK cells and a decline in their IFNγ and granzyme B production. Although an involvement of the purinergic pathway was not analyzed, the authors showed in a pathway enrichment analysis using the KEGG database differentially expressed genes for “nucleotide binding” and “ATP-binding” (Wang et al., 2023). Thus, we speculate that NK cells indeed can be negatively modulated by CD73/adenosine in the normal tissue and also that a therapeutic CD73 targeting could boost their activation and cytotoxic potential.
Besides a modulation of innate immune responses, also adaptive immune responses would be affected by an anti-CD73 therapy. We and others already highlighted that lymphocytes are increased in lung cancer patients and experimental mice and that recruited T lymphocytes release pro-inflammatory TNFα, IFNγ, IL-2, and lymphotactin during the early pneumonitic phase, which lasts from week 3–12 post radiation in a murine model (Schaue and McBride, 2012; Schaue et al., 2012; Wirsdörfer and Jendrossek, 2016; Lierova et al., 2018; Zhou P. et al., 2020). Following thoracic irradiation in mice, in addition to a typical Th1 response, a pro-inflammatory Th17-dominant
留言 (0)