
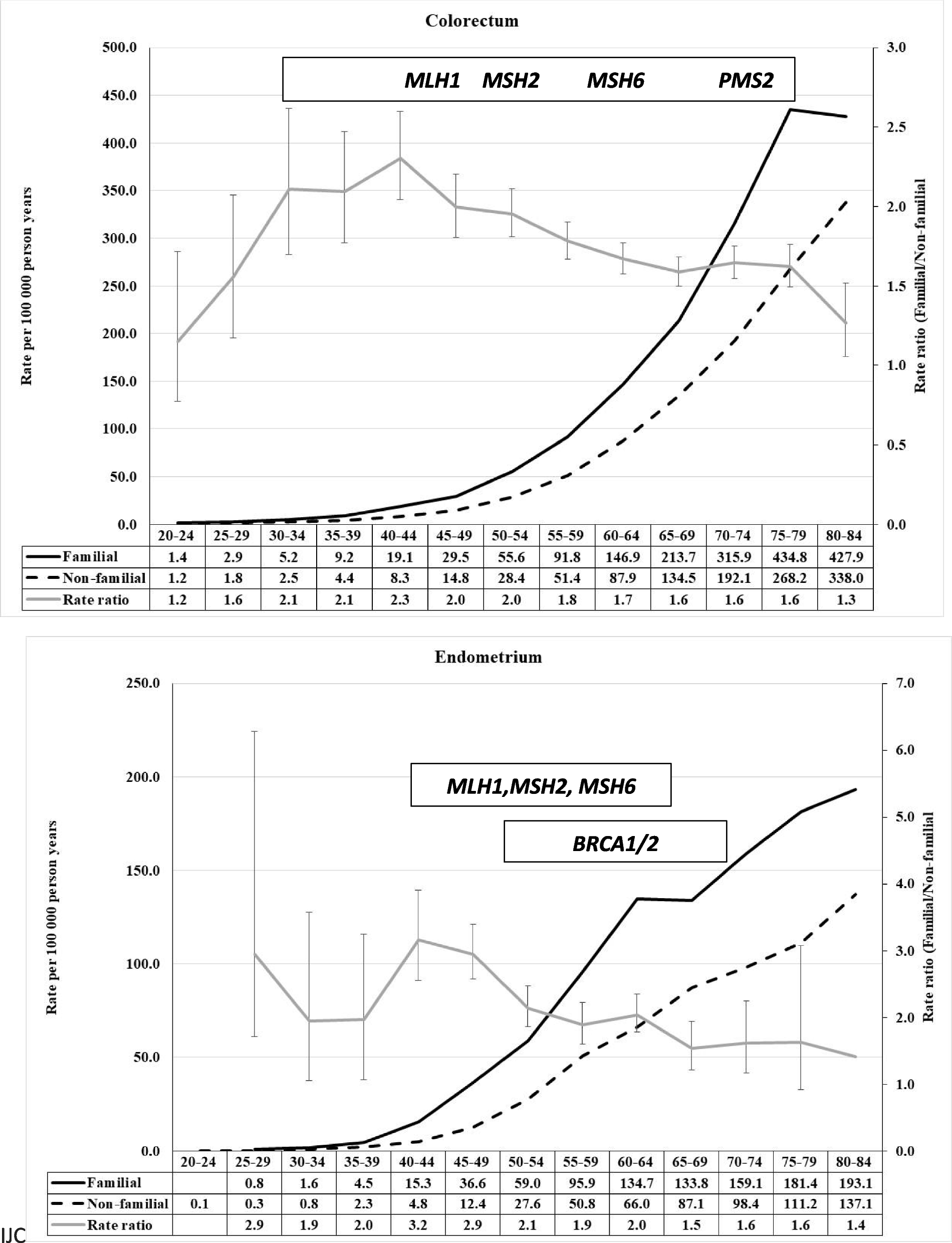
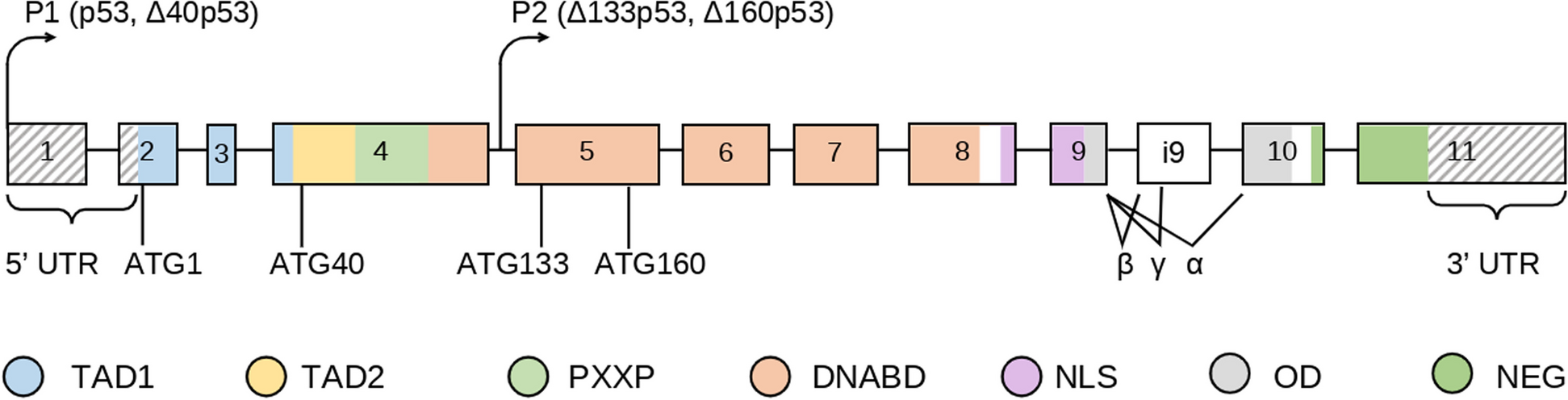
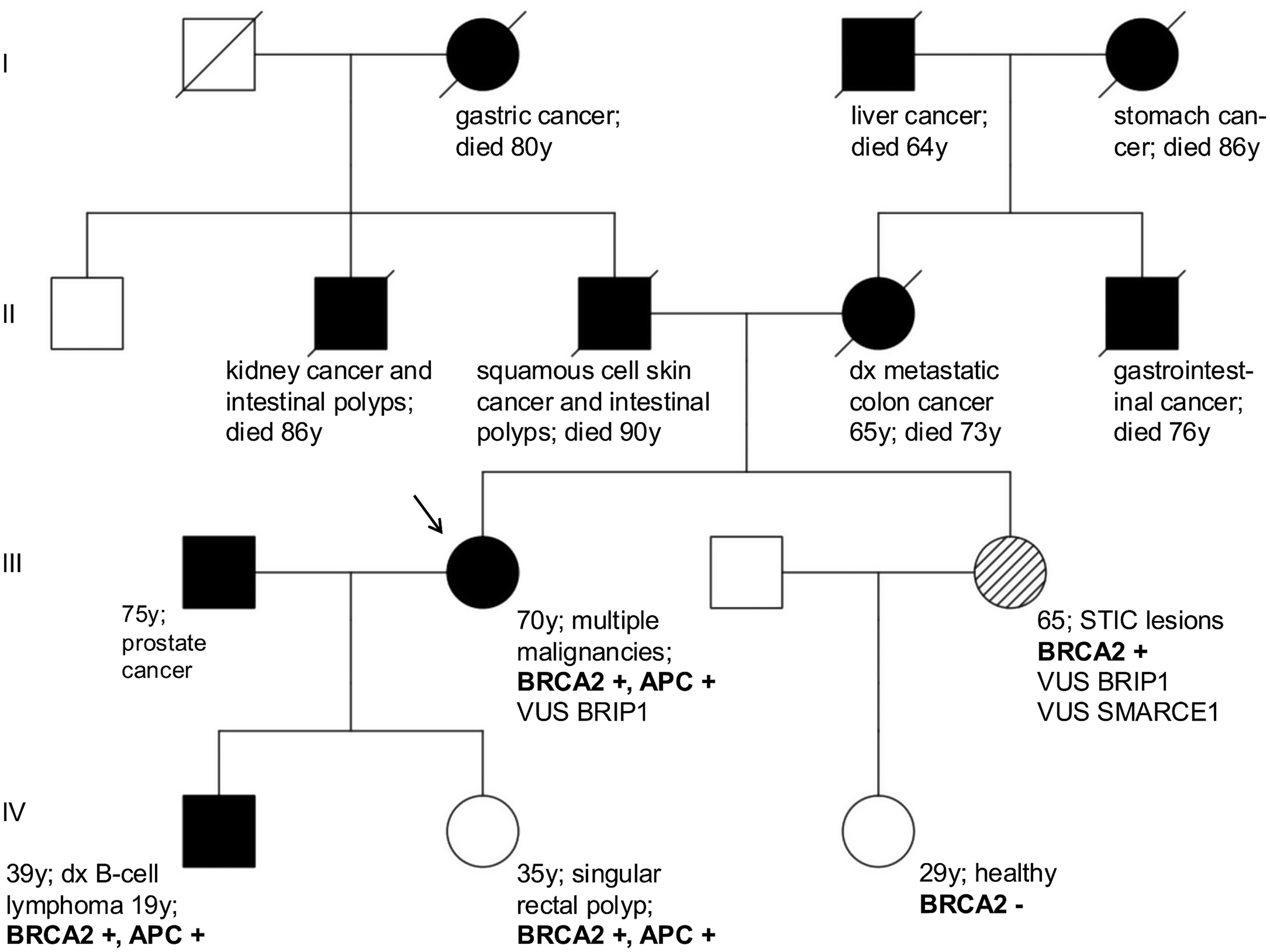
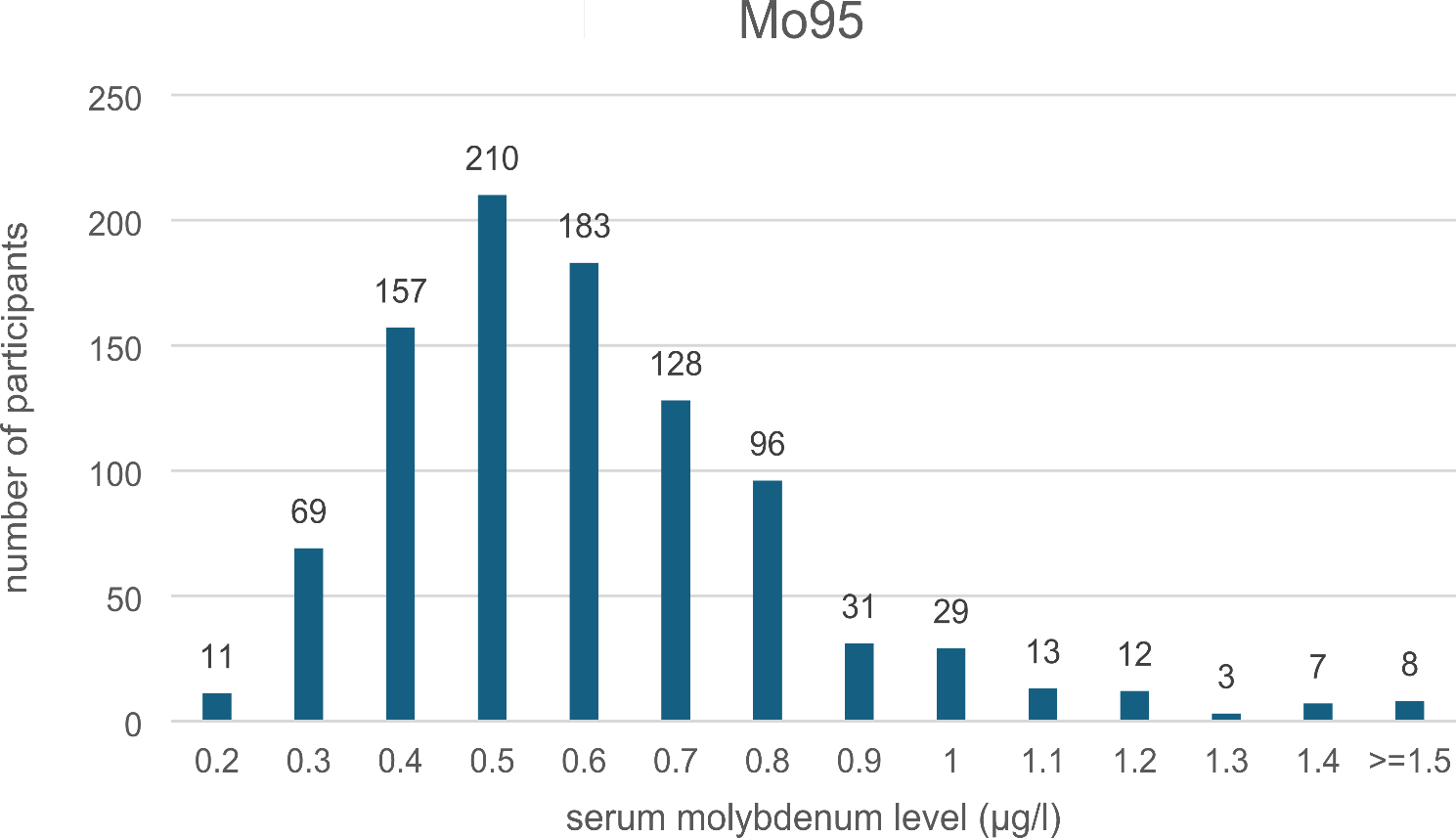
記住我






Patients with 5q heterozygous deletions exhibit various phenotypes that are not yet fully linked to a specific genetic cause. The most common clinical features include predisposition to cancer, ID, dysmorphic facies, and neurodevelopmental delay. Privitera et al. reported a case of FAP and ID with a stromal 19.85 Mb deletion in chromosome 5q, including the APC region and reviewed previous 12 cases reported or registered in the Decipher database [20]. The overlap of all the deleted regions was approximately cytobands 5q22.1q23.1 (7.77 Mb). In this study, we report two patients with ID, each from an independent family, with a large interstitial deletion at 5q21.2. q22.3 (Family 1), and 5q22.1. q23.1 (Family 2), which includes the APC region. In our study, we were able to narrow down the common deletion regions reviewed by Privitera et al.. 22 OMIM-registered genes (CAMK4, STARD4, STARD4-AS1, NREP, NREP-AS1, EPB41L4A, EPB41L4A-AS1, SNORA13, LOC101927023, EPB41L4A-DT, LINC02200, LOC102467216, APC, SRP19, REEP5, DCP2, MCC, TSSK1B, YTHDC2, KCNN2, LOC101927078, and LINC01957) were in the common deletion region (3.47 Mb) observed in our study for two probands (Fig. 4). Interestingly, the narrowed common region is completely included the region that Privitera et al. reviewed. Yamaguchi et al. reported a patient with FAP without ID who had a large genomic deletion in chromosome 5q22.1–22.2, which included CAMK4, STARD4, STARD4-AS1, NREP, NREP-AS1, EPB41L4A, EPB41L4A-AS1, SNORA13, LOC101927023, EPB41L4A-DT, LINC02200, LOC102467216, and APC [21]. Based on the findings of Yamaguchi et al. and our present report, we hypothesised that the following genes may cause ID when a large heterozygous deletion involving the APC region occurs: SRP19, REEP5, DCP2, MCC, TSSK1B, YTHDC2, KCNN2, LOC101927078, LINC01957. Among these, the genes with probability by gnomAD of being loss-of-function intolerant (pLI) of 0.9 or higher were YTHDC2 and KCNN2.
Fig. 4
Candidate genes that might cause intellectual disability in the case of heterozygous deletions. 22 OMIM-registered genes (CAMK4, STARD4, STARD4-AS1, NREP, NREP-AS1, EPB41L4A, EPB41L4A-AS1, SNORA13, LOC101927023, EPB41L4A-DT, LINC02200, LOC102467216, APC, SRP19, REEP5, DCP2, MCC, TSSK1B, YTHDC2, KCNN2, LOC101927078, and LINC01957) were in the common deletion region (3.47 Mb) observed in our study for the two probands. Yamaguchi et al. reported a patient with FAP with heterodeletions in 13 gene loci (CAMK4, STARD4, STARD4-AS1, NREP, NREP-AS1, EPB41L4A, EPB41L4A-AS1, SNORA13, LOC101927023, EPB41L4A-DT, LINC02200, LOC102467216, APC) [21]. Among the remaining nine genes, the genes with a probability of loss-of-function intolerance (pLI) due to gnomAD of 0.9 or higher were YTHDC2 and KCNN2. KCNN2 is reportedly associated with ID [22], while YTHDC2 is not
KCNN2 (MIM*605879) encodes the potassium calcium-activated channel subfamily N member 2, which is involved in membrane excitability. According to the OMIM database, this gene is associated with myoclonic dystonia type 34 (MIM#619724) and neurodevelopmental disorders, with or without various movement and behavioural abnormalities (MIM#619725), both of which have autosomal dominant inheritance. Intolerance to haploinsufficiency of KCNN2 is strongly supported by significant depletion in truncating variants of this gene in gnomAD [pLI = 0.99; putative loss-of-function observed/expected = 0.09 (0.04–0.27)]. Mochel et al. demonstrated that KCNN2 variants underlie dominant channelopathy, which is characterised by developmental delay and movement disorders [22]. Interestingly, all 11 patients with KCNN2 variants had an ID. YTHDC2 (MIM*616530) encodes an RNA helicase that is involved in RNA processing and metabolism. The intolerance of YTHDC2 to haploinsufficiency was supported by the significant depletion of truncating variants of this gene in gnomAD [pLI = 0.99; putative loss-of-function observed/expected = 0.06 (0.03–0.13)]. However, there are no reports associated with YTHDC2 and ID, nor has this been confirmed, even in the OMIM database. Based on current knowledge, this study confirmed that KCNN2 is most likely an autosomal dominant candidate gene for ID associated with 5q interstitial deletions.
In conclusion, we encountered two independent families with FAP with de novo heterozygous 5q deletions involving the APC region that could narrow down the 5p deletion region associated with ID. Although rare, patients with typical phenotypes of both FAP and ID require further investigation to understand ID caused by 5q stromal deletion.
留言 (0)