
記住我
Beta-herpesviruses (BHVs) infect a large proportion of the human population and are associated with a variety of pathophysiologic conditions (Crawford, 2023). Among BHVs, cytomegalovirus (CMV), also known as human herpesvirus 5 (HHV5 or HCMV), has received the most attention, and most knowledge about BHV has been gained through CMV research. Several approaches are currently being developed to combat HCMV infections, including an approach of targeting host cell proteins known as host-directed therapy (Kumar et al., 2020).
CMV infection completely reorganizes the infected cells and leads to many complex cytopathic effects, including the formation of two megastructures, the cytoplasmic assembly complex (AC) and the nuclear replication centers (Wofford et al., 2020; Sanchez and Britt, 2022; Turner and Mathias, 2022). The AC represents a part of the infected cell reorganized membrane system (ICRMS), which includes the relocation of the Golgi into a ring-like configuration that encloses a large perinuclear region containing early endosomes (EE), recycling endosomes (RE), the trans-Golgi network (TGN), and expanded membrane intermediates of the EE-RE/ERC-TGN interface (Das et al., 2007; Das and Pellett, 2011; Lučin et al., 2020). This structure, which is as large as the nucleus of the infected cell, is likely the site where the final steps of CMV virion assembly take place (Wofford et al., 2020), and it appears that the entire ICRMS contributes to virion egress (Wofford et al., 2020; Lučin et al., 2023). The majority of ER and LE membranes are extruded from the AC to the cell periphery (Beltran et al., 2016; Lučin et al., 2020).
Most studies on ICRMS have focused on the AC when this structure is fully established and the entire CMV gene repertoire is expressed, i.e. 96-120 h post-infection (hpi) in HCMV-infected cells (Das et al., 2007; Das and Pellett, 2011; Wofford et al., 2020). Reorganization is initiated early in infection and the basic configuration of the AC is established in the early (E) phase before viral DNA replication and late (L) gene expression (Sanchez et al., 2000; Das et al., 2007; Beltran et al., 2016; Rebmann GM et al., 2016; Procter et al., 2018; Taisne et al., 2019). In HCMV-infected cells, the establishment of the basic configuration takes 48 h or longer (Beltran et al., 2016; Wofford et al., 2020). However, the sequence of AC establishment has not yet been studied in detail and the developmental steps are poorly understood. Similar organizational changes occur in fibroblast-like cells infected with murine CMV (MCMV). In MCMV-infected cells, the AC is formed much earlier: the basic configuration is rapidly established between 5 and 7 hpi and evolves during the E phase of infection into a structure termed pre-AC (Taisne et al., 2019; Lučin et al., 2020). This is followed by the expression of numerous viral L genes that load pre-AC to establish AC, which matures further in the replication cycle, after viral DNA replication begins at 15–16 hpi (Lučin et al., 2020).
The membrane system reorganization is driven by viral gene products that target membrane-shaping regulatory host cell factors and thereby directly modulate their function (Das et al., 2014; Hook et al., 2014; Bughio et al., 2015; Dietz et al., 2018; Wu et al., 2020). This can also be achieved indirectly by stimulating their degradation (Lin et al., 2021) or by triggering signaling events that regulate membrane traffic (Cruz and Buchkovich, 2017). The early establishment of the basic configuration suggests an essential function of CMV E genes in the process, which actively remodel the membrane domains of the endosomal and secretory membrane system and reorganize membrane flux (Krzyzaniak et al., 2009; Tomaš et al., 2010; Hook et al., 2014; Zeltzer et al., 2018). Studies on HCMV-infected cells showed that several HCMV gene products, including pUL48 and pUL103 (Das et al., 2014), pUL97 (Azzeh et al., 2006), and gpUL132 (Wu et al., 2020), contribute to the formation of the ring-shaped configuration. Studies focusing on individual host cell factors that regulate the organization of membrane flux (Crump et al., 2003; Krzyzaniak et al., 2009; Tandon et al., 2009; Fraile-Ramos et al., 2010; Cepeda and Fraile-Ramos, 2011; Indran and Britt, 2011; Gudleski-O’Regan et al., 2012; Sharon-Friling and Shenk, 2014; Pavelin et al., 2017; Maschkowitz et al., 2018; Procter et al., 2018; Taisne et al., 2019; Hashimoto et al., 2020; Yang et al., 2022) and broader screening (McCormick et al., 2018; Turner et al., 2020) suggest that the virus targets diverse host cell machinery. The transcriptome studies showed that both HCMV (Hertel and Mocarski, 2004; Close et al., 2018) and MCMV (Juranic Lisnic et al., 2013; Lučin et al., 2020) affect the transcriptional activity of a large number of host cell genes related to membrane flux. In HCMV-infected cells (Weekes et al., 2014; Beltran et al., 2016; Jean Beltran et al., 2017a), many host cell proteins associated with membrane traffic are upregulated or downregulated, leading to an imbalance that may be associated with such extensive reorganization. This imbalance leads to an organization of membrane domains that differs from that observed in uninfected cells (Beltran et al., 2016; Tyl et al., 2022), and even to a distinct organelle structure (Jean Beltran et al., 2017b; 2017a; Zeltzer et al., 2018), which is associated with a displacement of many host cell proteins from the conventional organelles established in uninfected cells (Cook and Cristea, 2019). Therefore, it is becoming increasingly difficult to transfer the classical configuration of the membrane system established in uninfected cells to ICRMS. Accordingly, the physiology of membrane traffic is also likely to be altered and difficult to compare with that in uninfected cells. It is therefore essential to investigate the transport pathways in ICRMS in more detail and to compare them with the constantly growing knowledge about the membrane system of non-infected cells.
Studies on the part of the ICRMS known as the AC usually divide this structure into an outer AC (and the outer pre-AC in the E phase of infection) consisting of tangled and expanded Golgi elements surrounding the inner AC, which consists of expanded EE-, RE/ERC- and TGN-derived membrane structures in a large perinuclear area (Das and Pellett, 2011; Lučin et al., 2020; Wofford et al., 2020). The membrane structures within the inner AC are used for the final assembly of the CMV virions, the so-called secondary envelopment, and for the construction of an intracellular pathway for their exit from the cell (Wofford et al., 2020). Our studies on MCMV-infected cells have shown that unlinking and displacement of the Golgi and perinuclear expansion of the EE and REs/ERC domains are among the first cellular events that can be identified by immunofluorescence as signs of ICRMS and subsequent development of the AC (Lučin et al., 2020; Štimac et al., 2021). In the inner pre-AC, we first observed pericentriolar accumulation of membranes decorated with the small GTPase Rab10 (Karleuša et al., 2018; Lučin et al., 2020), and used it as an indicator of the earliest events of pre-AC biogenesis (Marcelić et al., 2021; Pavišić et al., 2021; Štimac et al., 2021; Štimac et al., 2024). However, little biochemical evidence has been provided for its enhanced membrane mobilization.
Rab10 is a versatile Rab that can be activated at different cellular localizations and can be involved in various processes at different developmental stages of the membrane system (Chua and Tang, 2018). Although Rab10 can be activated at the ER (Babbey et al., 2006; English and Voeltz, 2013; Liu et al., 2013), the ER is excluded from the inner pre-AC (Sanchez et al., 2000; Das and Pellett, 2011; Lučin et al., 2020), and the pericentriolar accumulation of Rab10 indicates the expansion of tubular intermediates at EEs or within the ERC (Karleuša et al., 2018; Lučin et al., 2020; Štimac et al., 2021). Rab10 can be activated on Rab5-positive EEs, where Rab5 and Rab10 can form a regulatory cascade in which Rab5 recruits the GDP-GTP exchange factor (GEF) to activate Rab10 (Liu et al., 2018), and Rab10 recruits the GTP hydrolysis activating protein (GAP) that inactivate Rab5 (Liu and Grant, 2015). Rab10 can also be activated simultaneously with Rab11 at EE as they can share the GEF (Xiong et al., 2012), which promotes the development of domains associated with the sorting of different endocytic cargo. Activation of Rab10, in cooperation with Rab22a, drives the development of tubular recycling endosomes (TREs) (Etoh and Fukuda, 2019; Farmer et al., 2021), which sort and recycle clathrin-independent endocytic cargo (CIE) to the PM (Xie et al., 2016). In cells forming TREs, Rab10 is recruited together with EHBP1 to phosphatidylinositol (4,5)bisphosphate (PI(4,5)P2)-enriched membranes (Farmer et al., 2021). The recruitment of Rab10 to TREs is facilitated by the recruitment of the Rab10 effector EHBP1 (Farmer et al., 2021). It appears that in most cells, the established Rab10 domain at the EEs is highly dynamic, the generated tubules are rapidly converted into transport intermediates and Rab10 is rapidly removed from the membranes and released into the cytosol (Babbey et al., 2006). Rab10 function may also be associated with the downstream population of tubular membranes that process and recycle CIE cargo known as Arf6/Rab8-REs (Kobayashi and Fukuda, 2013). However, Rab10 can also be activated downstream in the recycling tract, at a subset of Rab11-REs, by Rab11, which recruits Rabin8, GEF for Rab8 and Rab10 (Homma and Fukuda, 2016).
The Rab10-positive domain (Rab10-PD) in the inner pre-AC of MCMV-infected cells may be co-opted by the infection for the needs of the CMV replication cycle and could be a biomarker for pre-AC biogenesis. Therefore, it would be important to determine whether Rab10 is over-recruited to membranes and Rab10-PD expanded within the inner pre-AC. To do so, we performed long-term live imaging of GFP-Rab10 using digital holotomographic microscopy (DHTM) in combination with epifluorescence, confocal imaging of known Rab10 interactors and identification of important Rab10 interactors using the BioID (proximity-dependent biotin identification) assay (Roux et al., 2012). We also analyzed perinuclear Rab10 accumulation after knock-down of EHBP1 and Rabin8, two proteins required for Rab10 recruitment to membranes (Farmer et al., 2021), and after blocking PI(4,5)P2-rich membrane domains. Our study demonstrates the gradual expansion of Rab10-PD in the inner pre-AC as Rab10 associates with EHBP1 and MICAL-L1. The formation of the expanded Rab10-PD requires EHBP1 and PI(4,5)P2, but not Rabin8. These data suggest the expansion of a subset of EE-derived TRE-like membranes in pre-AC, which may serve as the earliest biomarker of pre-AC biogenesis.
2 Materials and methods2.1 Cell lines and cell cultureThe NIH 3T3 (ATCC CRL-163) and Balb 3T3 (clone A31, ATCC CCL-163) murine fibroblast-like cell lines were propagated in 10 cm Petri dishes and divided into appropriate plates once they were 80%–90% confluent. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 mg/mL streptomycin and 100 U/mL penicillin (all reagents from Gibco/Invitrogen, Grand Island, NY, United States) at 37°C and 5% CO2.
2.2 Murine cytomegalovirus and infection proceduresFor infection, we used Δm138-MCMV (ΔMC95.15), a recombinant virus with the deletion of the viral fcr1 (m138) gene (Crnković-Mertens et al., 1998). In some experiments, we used the MCMV wild-type strain Smith (ATCC VR-194; American Type Culture Collection [ATCC]). The production of MCMV stocks and infection conditions were according to the standard procedures as described before (Brizić et al., 2018). In brief, adherent cells were infected with 1 PFU/cell, and a multiplicity of infection (MOI) of 10 was achieved by centrifugal enhancement (2000 rpm/15 min). Immediate-Early-1 protein (pIE1) monitoring by immunofluorescence or Western blot was performed regularly to verify infection as previously described (Lučin et al., 2020).
2.3 Antibodies and reagentsThe following monoclonal (mAb) or polyclonal (pAb) antibodies against host cell proteins were used in this study: rabbit mAb (IgG) to Rab10 (Cat.No. 8127; Cell Signaling Inc, Danvers, MA, United States), rabbit pAb (IgG) to Rab3IP/Rabin8 (Cat.No. 12321-1-AP; Proteintech, Manchester, United Kingdom), rabbit mAb (IgG) to Rab5 (Cat.No. 3547; Cell Signaling Inc, Danvers, MA, United States), rabbit pAb (IgG) to TBC1D4/AS160 (Cat.No. LS-C368534; LS-Bio, Shirley, MA, United States), rabbit pAb (IgG) to TBC1D2 (Cat.No. LS-C157145; LS-Bio, Shirley, MA, United States), rabbit pAb (IgG) to EHBP1 (Cat.No. 17637-1-AP; Proteintech, Manchester, United Kingdom), rabbit pAb (IgG) to ACAP1/CENTB1 (Cat.No. orb182538; Biorbyt, Cambridge, United Kingdom), mouse mAb (IgG) to ACAP2/CENTB2, (Cat. No. Sc-376150; Santa Cruz Biotechnology, Dallas, United States), rabbit pAb (IgG) to MICALL1 (Cat.No. orb537847; Biorbyt, Cambridge, United Kingdom), mouse mAb (IgG1) to GM130 (Cat. No. 610823, BD Biosciences, Franklin Lakes, NJ, United States), and mouse mAbs to β-actin (IgG1; Cat. No. MAB150; Millipore, Burlington, Massachusetts, United States) and to HA (Cat.No. 51064-2-AP; Proteintech, Manchester, United Kingdom). W6/32 mAb (mouse IgG2a, ATCC HB-95) that react with human but not with mouse MHC class I proteins was used as hybridoma supernatant. Monoclonal antibodies against MCMV protein, I.E.,1 (pm123/pIE1) included clones CROMA101 (IgG1) and, I.E.,1.01 (IgG2a), produced by the University of Rijeka Center for Proteomics, (https://products.capri.com.hr/shop/?swoof=1&pa_reactivity=murine-cytomegalovirus; accessed on 25 October 2024).
For immunofluorescence analysis, we used Alexa Fluor (AF)-conjugated (AF488, AF594, and AF555) secondary Abs to mouse IgG2a, mouse IgG1, and rabbit IgG (Molecular Probes, Leiden, Netherlands), and AF680-conjugated secondary Abs to IgG1 and IgG2a (Jackson Laboratory, Bar Harbor, ME, United States). Streptavidin-AF488 was from Thermo Fisher Scientific (Waltham, MA, United States). For Western blot analysis, we used HRP-conjugated goat anti-rabbit and goat anti-mouse Abs (Jackson Laboratories, Bar Harbor, ME, United States). DAPI (4,6-diamidino-2-phenylindole dihydrochloride) was obtained from Thermo Fisher Scientific (Waltham, MA, United States; Cat. No. D1306), puromycin from Santa Cruz Biotechology Inc (Dallas, United States), and propidium iodide and biotin from Sigma-Aldrich Chemie GmbH (Schnelldorf, Germany).
2.4 Immunofluorescence and confocal microscopyThe 60%–70% confluent cells grown on coverslips in 24-well plates were MCMV infected, fixed in 4% paraformaldehyde (PFA) for 20 min at room temperature (r.t.), and permeabilized in 0.5% Tween 20 for 20 min at 37°C. Primary Abs and AF-conjugated secondary Abs were incubated for 60 min at 37°C, the excess of Abs was washed three times with T-TBS, and samples were embedded in Mowiol (Fluka Chemicals, Selzee, Germany)-DABCO (Sigma Chemical Co, Steinheim, Germany) in PBS containing 50% glycerol. The samples were analysed by Leica DMI8 inverted confocal microscope (confocal part: TCS SP8; Leica Microsystems GmbH, Wetzlar, Germany), equipped with HC PLAPO CS2 objective (×631.40 oil), four lasers (UV with Diode 405 for DAPI; Ar 488 for AF488; DPSS 561 for AF555 and AF595; and He/Ne 633 for AF680), and four detectors (two of which are PMT and two are HyD). Images were acquired in sequential mode (515 × 515 pixels, z-series of 0.5 μm) using LAS (Leica Application Suite) X version 3.5.6.21594 software (Leica Microsystems GmbH, Wetzlar, Germany). The offset was set to 0%–1.5% depending on the background. All samples that were compared within an experiment were imaged with the same parameters.
2.5 Time-lapse acquisitions using holotomographic microscopyHolotomographic microscopy in combination with epifluorescence was performed using the 3D Cell-Explorer Fluo (Nanolive, Ecublens, Switzerland) with a ×60 air objective at a wavelength of λ = 520 nm. The irradiance of the laser was 0.2 nW/μm2, and the acquisition time per image was 45 m. An top-stage incubator (Oko-lab, Pozzuoli, Italy) equipped with a heatable glass lid to avoid condensation was used to maintain physiological conditions during live cell imaging, including a constant temperature of 37°C, 90% relative humidity and 5% CO2 concentration. For imaging, 50,000 cells infected with Δm138-MCMV (MOI 10) were plated in 35-mm glass-bottomed Ibidi dishes (Ibidi GmbH, Germany). The dishes were placed in the incubation chamber for acclimatization prior to imaging. Imaging started 6 h after infection. The refractive index image was acquired every 2.5 min for 11 h in combination with epifluorescence every second frame. The time-lapse images were saved as 3D stacks using STEVE software (Nanolive SA, Ecublens, Switzerland), which controls the 3D Cell Explorer microscope. The 3D volumes were converted to 2D maximal projections along the z-axis. The 3D stacks obtained with DHTM were digitally stained with STEVE, divided into voxel segments and exported as TIFF files.
2.6 Image analysisPericentriolar (pc) and perinuclear (pn) accumulation of Rab10 fluorescence signal in MCMV-infected cells within an angle of α ≤ 90° was defined as pre-AC as previously described (Štimac et al., 2021). The pre-AC-positive cells were counted on at least 10 fields of view using an Olympus BX51 epifluorescence microscope equipped with a DP71CCD camera (Olympus, Tokyo, Japan) and a UPlanFL N 40×/0.75 objective.
Colocalization was determined using Manders’ overlap coefficients (M1 and M2) calculated on the entire z-stack of confocal images (at least 8–12 slices; 120.37 × 120.57 nm pixel size) using the Fiji-ImageJ and the JACoP plugin (https://imagej.net/ij/plugins/track/jacop2.html, accessed on 25 October 2024) (Bolte and Cordelières, 2006). After splitting the red, green and blue channels and subtracting the background, the colocalization of the pixels between two selected channels was determined as previously described (Marcelić et al., 2022).
The DHTM images exported in TIFF format from STEVE were processed in Fiji-ImageJ. The overlaid images with RI and fluorescence signals were split, the cell area and the fluorescence area above the background were delineated with the freehand tool and used for measurement, including the surface area and average fluorescence intensity. The cell area was calculated on the RI image through the focal plane and the Rab10-positive area was calculated on the fluorescence image using the freehand tool and the region of interest (ROI) manager. The average fluorescence intensity of the Rab10-positive area was calculated on the fluorescence image using the ROI manager.
2.7 siRNA silencingSmall interfering (si)RNA sequences were acquired as follows: negative (control) siRNA (1022076) was from Qiagen (Hilden, Germany); siRNA for Rab3IP/Rabin8 (Sc-152666) and siRNA for EHBP1 (Sc-144602) were from Santa Cruz Biotechnology Inc. (Dallas, United States); and siRNA for Rab10 (L-040862-01–0005) was from Dharmacon Inc. (Laffayete, Colorado, United States). Briefly, siRNA and RNAiMAX Lipofectamine Reagent (Invitrogen, Carlsbad, CA, United States) were mixed according to the manufacturer’s guidelines, added dropwise to the cells, incubated at 37°C for 48 h, and analysed or proceeded to the experiment. The final concentration of siRNA for Rab3IP/Rabin8 was 60 nM, for EHBP1 was 100 nM, and for Rab10 was 30 nM.
2.8 Western blotCells were lysed in RIPA buffer containing protease inhibitors (Cat. No. 11697498001, Roche Diagnostics GmbH, Unterhaching, Germany), the lysates were mixed with the sample buffer, the proteins were separated by SDS-PAGE using Bio-Rad PowerPac (Universal, Hercules, CA, United States) and blotted onto a polyvinylidene (PVDF-P) difluoride membrane (Millipore, XXX) using the Bio-Rad Trans-Blot Turbo Transfer System (Hercules, CA, United States) at 60–70 V for 1 h. Membranes were blocked for 1–2 h in 1% blocking reagent (Roche Diagnostics GmbH, Mannheim, Germany), incubated overnight at 4°C with primary Abs, washed three times in T-TBS (TBS with 0.05% Tween 20; pH = 7.5) and incubated for 45–60 min with peroxidase (POD)-conjugated secondary Abs in T-TBS with 0.5% blocking reagent. After washing in T-TBS, the signal was detected by chemiluminescence (SignalFire [TM] Plus ECL Reagent or SignalFire [TM] Elite ECL Reagent; Cell Signaling, Cat. No. 12630S and 12757P, respectively) using Transilluminator Alliance 4.7 (Uvitec Ltd., Cambridge, United Kingdom) and ImageQuant LAS 500 (GE Healthcare Bio-Sciences AB, Upsala, Sweden). In each experiment, β-actin was used as a loading control and detected on the same membrane. The chemiluminescence signal was quantitatively analyzed using ImageQuantTL (version 10.2., Cytiva) and ImageJ 1.53 software. All values were normalized to the signal of β-actin by calculating the lane normalization factor as follows: observed actin signal for each lane/highest observed actin signal for the blot. The value normalized to actin was then used to normalize the experimental signal (raw signal value/normalized actin index). The kinetics of host cell protein expression during MCMV infection was calculated as = normalized experimental signal (tx hpi)/normalized experimental signal (t0 hpi).
2.9 Subcloning of EGFP-Rab10wt and EGFP-Rab10Q68L into the lentiviral vector pLIX-Kan_PstIEGFP-Rab10wt and EGFP-Rab10Q68L ORFs were subcloned from EGFP-Rab10 (Rzomp et al., 2003) and EGFP-Rab10-Q68L (Huang et al., 2010) plasmids (a gift from dr. Marci Scidmore; Addgene plasmids # 49742 and # 49544) into the pLIX402_Kan lentiviral vector with doxycycline-inducible expression of transgene derived from pLIX402 (Addgene plasmid # 41394) by insertion of Kanamycine kassete (a gift from dr. Martin Messerle (Hannover, Germany) (Kutle et al., 2020). Briefly, the primers for PCR amplification of bothORFs (F-EGFP-Rab10: 5′-TTT TTT CTG CAG ATG GTG AGC AAG GGC GAG-3′, and R-GFP-Rab10: 5′-TTT TTT GGA TCC TCA GCA TTT GCT CTT CC-3′) were created according to original plasmids. At the same time, the pLIX-Kan vector was cleaved with PstI (cat. no. R3140S, New England Biolabs Inc.) and BamH1 (cat. no. R3136S, New England Biolabs Inc.) restriction endonucleases. Finally, rapid DNA ligation kit (cat. no. K1422, Thermo Fisher Scientific, Waltham, MA, United States) was used to ligate EGFP-Rab10 insert into pLIX lentiviral vector (without Kan cassette).
The verified pLIX EGFP-Rab10wt and EGFP-Rab10Q68L plasmids were used for production of lentiviruses in HEK 293T cells (ATCC clone A31, ATCC CRL-3216, Manassas, VA, United States). Transduction of NIH 3T3 cells and selection of NIH 3T3 EGFP-Rab10wt and NIH 3T3 EGFP-Rab10Q68L cells with puromycin (2.5 μg/mL) is described in Section 2.11.
2.10 Construction of the pGenLenti Rab10-BioID2-HA plasmidRab10-(GGGGS)13-mBioID2-HA fusion sequence was designed in silico by in-frame joining of mouse Rab10 protein-coding sequence (retrieved from Ensembl genome database, ensesmbl. org), 13×(GGGGS) linker sequence (Kim et al., 2016), mouse codon-optimized BioID2 (mBioID2) sequence, and a sequence encoding HA-tag. BioID2 sequence was retrieved from the plasmid MCS-BioID2-HA (a kind gift from Kyle Roux, n2t.net/addgene:74224), and subsequently codon-optimized for expression in mouse cells using GenSmart™ Codon Optimization tool available at www.genscript.com/gensmart-free-gene-codon-optimization.html). Once designed, the Rab10-(GGGGS)13-mBioID2-HA fusion sequence (see supplementary material and methods) was submitted to GenScript, where the corresponding DNA fragment was synthesized and subcloned into the pGenLenti lentiviral vector (GenScript Biotech., New Jersey, United States) between EcoRI and BamHI restriction sites, giving rise to the pGenLenti Rab10-BioID2-HA plasmid. Purified pGenLenti Rab10-BioID2-HA was then used to produce lentiviral particles in HEK 3T3 cells (ATCC clone A31, ATCC CRL-3216, Manassas, VA, United States). Obtained lentiviral particles carrying Rab10-BioID2-HA were then used to transduce NIH 3T3 cells, and Rab10-BioID2 positive NIH 3T3 cells were selected using puromycin (2 μg/mL).
2.11 Generation of NIH 3T3 EGFP-Rab10wt, NIH 3T3 EGFP-Rab10Q68L and NIH 3T3 Rab10-BioID2-HA cell linesNIH 3T3 cells expressing EGFP-Rab10 or Rab10-BioID2-HA fusion proteins were generated by transduction with lentiviruses. 5 μg of the lentiviral plasmid pLIX EGFP-Rab10wt, pLIX EGFP-Rab10Q68L or pGenLenti Rab10-BioID2-HA were mixed with 10 μg of p8.91 (a gift from Simon Davis; Addgene plasmid # 187441; http://n2t.net/addgene:187441; RRID:Addgene_187441) (Sušac et al., 2022) and 0.5 μg of plasmid p-CMV-VSV-G (a gift from Bob Weinberg; Addgene plasmid #8454; http://n2t.net/addgene:8454; RRID:Addgene_8454) (Stewart et al., 2003), dissolved in 1.5 mL Optimem (Thermo Fisher Scientific, Waltham, MA, United States; (Cat. No. 31-985-070). The solution was mixed with 41 μL Lipofectamine 3000 in 1.5 mL Optimem and carefully added to 80%–85% confluent HEK 393T cells (ATCC clone A31, ATCC CRL-3216, Manassas, VA, United States) cultured in 10% FCS DMEM without antibiotic. The supernatants were collected 24, 30 and 48 h after transfection. After centrifugation (5 min at 2,000 rpm) and filtration (0.45 μM filter), the lentiviruses were used to transfect NIH 3T3 cells. Puromycin (2.5 μg/mL) was used to select the fusion protein-expressing cells. The puromycin-resistant EGFP-Rab10-expressing cells were additionally sorted using the FACSAria cell sorter (Becton Dickinson and Co, San Jose, CA, United States).
2.12 Biotin labelling of NIH 3T3 Rab10-BioID2-HA cells and immunoprecipitation of biotinylated proteinsNIH 3T3 Rab10-BioID2-HA cells were cultured in 6 cm Petri dishes in cell culture medium with biotin (50 μM) as follows: (1) 18 h for uninfected cells; (2) 12 h before infection and 6 h together with MCMV infection (without replacement of the culture medium), and (3) 18 h together with MCMV infection. After three washes with PBS to remove excess biotin, 20% of cells were lysed in 20 μL of RIPA to achieve whole cell lysate (WCL), and 80% of the cells were lysed in 350 μL of RIPA for overnight precipitation of biotinylated proteins with NeutrAvidin Agarose (NA-A) (Thermo Fisher Scientific, SAD) at 4°C. The samples were centrifuged (14000 rpm, 1 min), and the pellet was twice washed in washing buffer 1 (1% SDS), then once in washing buffer 2 (0.1% deoxycholic acid, 1% Triton-X100, 1 mM EDTA, 500 mM NaCl, 50 mM HEPES), and washing buffer 3 (0.5% deoxycholic acid, 0.5% NP-40, 1 mM EDTA, 250 mM LiCl, 10 mM Tris-HCl pH 7.4), as described (Sakai et al., 2019; Nguyen-Tien et al., 2022). The biotinylated proteins were eluted for 10 min at 95°C in a buffer containing 10% 2-mercaptoethanol, 0.125 M Tris-HCl (pH 6.8), 0.1% SDS (w/v), 0.1% bromophenol blue (w/v), and 30% glycerol (v/v) followed by Western blot analysis together with WCL. The pIE1 served as control of infection, and β-actin as loading control.
2.13 Transient transfection of NIH 3T3 and Balb 3T3 cellswith EGFP-Rab10, EGFP-Rab10-Q68L, and EGFP-PH-PLC-δ1 MSCV plasmidsThe original Addgene plasmids EGFP-Rab10 (Rzomp et al., 2003) and EGFP-Rab10-Q68L (Huang et al., 2010) were used for transfection to express the wild-type and GTP-locked forms of Rab10, respectively. Murine Stem Cell retroviral vector (MSCV) containing EGFP-PH-PLC-δ1 (pleckstrin homology domain from phospholipase C delta 1) was used to express the recombinant membrane domain that binds to PI(4,5)P2 (Bertović et al., 2020). Transient transfections was performed by gently mixing solutions of 1 μg of plasmids in Optimem (Gibco/Invitrogen, Grand Island, NY, United States) with 1 μL of Lipofectamine 3000 transfection reagent (Invitrogen) according to the manufacturer’s instructions and proceeded to the experimental protocols as described in Results.
2.14 Statistical analysis and data presentationStatistical significance was assessed using a two-tailed Student’s t-test. Differences were considered significant when P values were <0.05 (*P ≤ 0.05; **P < 0.01; ***P < 0.001).
3 Results3.1 Expansion of Rab10-positive domain within the reorganized membrane system of cytomegalovirus-infected cellIn our previous studies on Balb 3T3 cells (Lučin et al., 2020; Štimac et al., 2021; 2024), we reported the perinuclear accumulation of Rab10-positive membranes in Δm138-MCMV-infected cells in the E phase of infection. Very little endogenous Rab10 can be detected on membrane structures in these cells (Figure 1A, uninfected). After infection, perinuclear accumulation of Rab10 was observed by confocal imaging in 32.8% ± 2.8% of infected cells at six hpi, which progressed during the E phase of infection and resulted in 72.0% ± 3.3% of infected cells showing increased perinuclear Rab10 staining at 16 hpi, at the end of the E phase (Figures 1A, B). Nuclear accumulation of immediate early one (pIE1) was used as an indicator of infection (Figure 1A). For infection, we used recombinant MCMV with deleted m138 gene encoding the E-phase protein with FcR properties (Thäle et al., 1994) to visualize endogenous host cell proteins and avoid nonspecific binding of antibody reagents in immunofluorescence studies. Infection with the wild type virus, expressing m138 protein with Fc-binding properties (Supplementary Figure S1A), resulted in indistinguishable reorganization of the membrane system in the E phase of infection, including dislocation of the Golgi (Supplementary Figure S1A) and perinuclear accumulation or Rab10 (Supplementary Figure S1B).
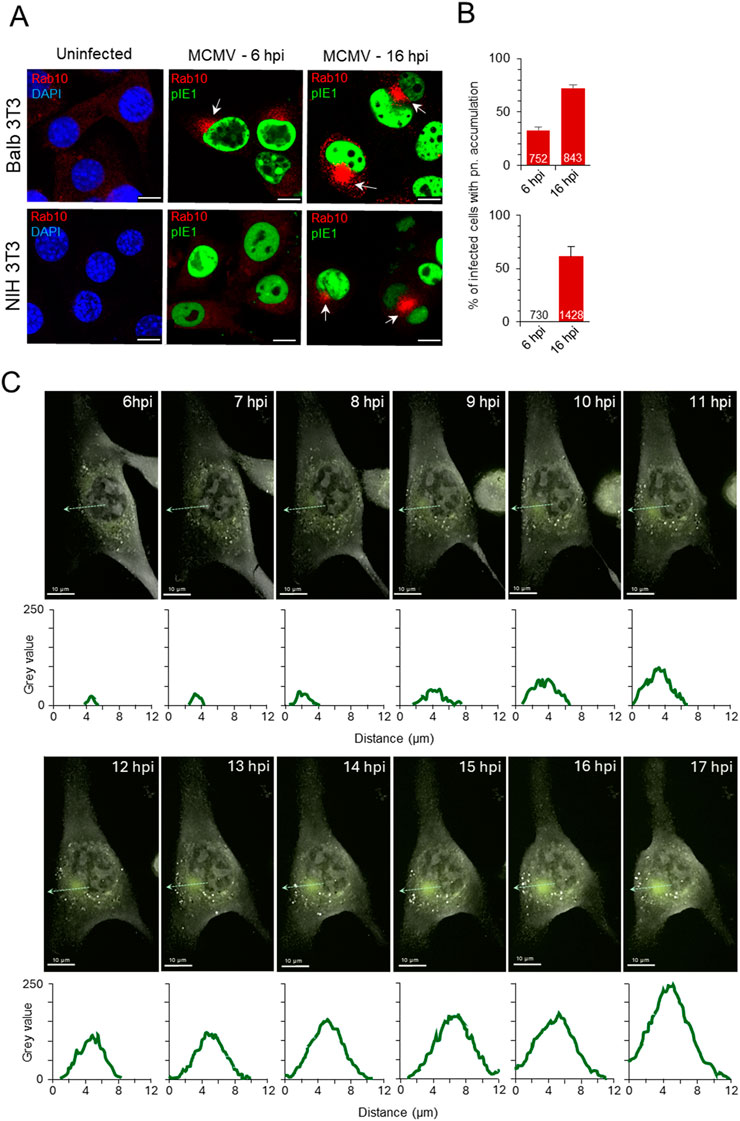
Figure 1. Expansion of Rab10-PD in the pre-AC of CMV-infected cells. (A, B) Balb 3T3 and NIH 3T3 cells, uninfected or infected with Δm138-MCMV (MOI of 10), were fixed 6 and 16 h post-infection (hpi), permeabilized and stained with Abs against Rab10 (red) in combination with Abs against pIE1 in infected cells for infection control (green) or with DAPI (blue) in uninfected cells for nuclei staining. Shown are the merged confocal images through the focal plane. The arrows show the perinuclear accumulation of Rab10 in the pre-AC. Bars, 10 μm. The percentage of cells (B) in three independent experiments with perinuclear (pn) Rab10 accumulation in MCMV-infected (pIE1-positive) cells is shown as mean ± SD. The numbers in the bars indicate the total number of cells analyzed. (C) Long-term high-frequency live cell imaging of MCMV-infected cells using DHTM with attached epifluorescence module. Expression of EGFP-Rab10 in the NIH 3T3 EGFP-Rab10 cell line was induced by doxycycline (2 μg/mL), and after 24 h, cells were infected with Δm138-MCMV (MOI of 10). Cells were imaged continuously from six hpi with DHTM at intervals of 2.5 min for refractive index (RI) and 5 min for fluorescence signal. The overlaid images show RI and fluorescence signal at 1-h intervals during the E phase of infection (6–17 hpi). The RI and fluorescence images as well as the overlaid video can be seen in the supplementary (Supplementary Video S1). The fluorescence intensity profiles along the light green dashed arrow lines are shown below the images, and the surface area and mean fluorescence intensity of the Rab10-positive perinuclear region is shown in Supplementary Figure S3. Another example of the cell is shown in Supplementary Figure S4 and Supplementary Video S2.
Similar to Balb 3T3 cells, Rab10 was barely detected in cytoplasmic structures of uninfected NIH 3T3 cells (Figure 1A). After MCMV infection a similar proportion of cells (61.3% ± 9.3%) developed enhanced perinuclear Rab10 accumulation at 16 hpi, but no perinuclear Rab10 accumulation was observed at six hpi (Figures 1A, B). Apart from the observed delay in Rab10 accumulation, the development of pre-AC in NIH 3T3 cells is consistent with previously published data on Balb 3T3 cells (Lučin et al., 2020; Štimac et al., 2021), including the rapid turnover of activated Rab10 at endosomal membranes of the uninfected cell.
To investigate the expansion of Rab10-PD in living cells, we generated cell lines on the NIH 3T3 background with inducible expression of wild-type and GTP-locked (Q68L) mutant of Rab10 (NIH 3T3 EGFP-Rab10wt and NIH 3T3 EGFP-Rab10Q68L, respectively), as confirmed by fluorescence imaging (Supplementary Figure S2A) and Western blot (Supplementary Figure S2B). A modest organelle-associated fluorescent signal was detected in these cells after 24 h of induction, mainly in the pericentriolar region (Supplementary Figure S2A). We infected these cells with MCMV 24 h after induction of EGFP-Rab10wt expression and performed long-term time-lapse imaging during the early phase of infection. We combined digital holo tomographic microscopy (DHTM), which provides high-resolution refractive index (RI) images at high spatial resolution with ultra-low power, with epifluorescence time-lapse imaging of the fluorescence signal. Infected cells were placed into the top-stage incubator immediately after infection, and DHTM recording began 2–3 h later at high frequency (every 2.5 min for refractive index and every 5 min for fluorescence) over the next 16–17 h. Several cytopathic effects were observed during DHTM recording, including nuclear rotation as described for HCMV-infected cells (Procter et al., 2018) (Supplementary Video S1). At 6–7 hpi, nuclear indentation began in the pericentriolar region, accompanied by the concentration of fluorescence signal as a pericentriolar spot at eight hpi (Figure 1C; Supplementary Figure S3; Supplementary Video S1). As the E phase progressed (from 8 to 17 hpi), the surface area and fluorescence in the perinuclear region increased (Figures 1C; Supplementary Figure S3, S4; Supplementary Videos S1, S2), demonstrating the expansion of Rab10-PD. Similar perinuclear accumulation was observed by live imaging of MCMV-infected cells expressing GTP-locked Rab10 (Supplementary Video S3) and in cells transfected with EGFP-Rab10Q68L (Supplementary Figure S5), indicating membrane recruitment of GTP-bound form of Rab10 at membranes within the pre-AC. We also confirmed EGFP-Rab10wt accumulation in the E phase after infection with the wild-type MCMV (Supplementary Video S4).
3.2 Accumulation of EHBP1, Rabin8 and Rab10 effectors in the inner pre-ACAlthough Rab10 can interact with many host cell proteins associated with the regulation of membrane flux in the secretory and endosomal system of the cell (Chua and Tang, 2018; Zhu et al., 2024), its perinuclear accumulation in CMV-infected cells suggests membrane expansion at the EE-RE/ERC-TGN interface (Lučin et al., 2020). Therefore, we performed immunofluorescence analysis of the known interactors of Rab10 at EE and the ERC (Figure 2A). As previously reported in Balb 3T3 cells (Lučin et al., 2020), we observed perinuclear accumulation of several small GTPases that act at the EE-ERC interface and are associated with Rab10 activation, including Rab5 and Rab11. Similar was observed in NIH 3T3 cells (data not shown). As described for Rab10, six of the Rab10 interactors analyzed in uninfected NIH 3T3 cells showed no staining of membrane organelles, with the exception of Rabin8, which was detected on various structures at the cell periphery (Figure 2B). In MCMV-infected NIH 3T3 cells, endogenous EHBP1, a protein essential for the recruitment of Rab10 to EEs (Farmer et al., 2021), accumulated in the perinuclear area at 16 hpi (Figure 2B) in 62.4% ± 6.1% of infected cells (Figure 2C), similar to Rab10 (Figure 1B). Endogenous Rabin8, an effector of Rab11 that functions as a GEF for Rab10 and Rab8 downstream in the ERC (Homma and Fukuda, 2016), accumulated mainly at the cell periphery and to some extent in the perinuclear area (Figure 2B) of 23.8% ± 7.3% of infected cells at 16 hpi (Figure 2C). The Rab10 effectors TBC1D2 (GAP for Rab5 (Liu and Grant, 2015), and AS160 (known as TBC1D4; GAP for Rab10 (Homma et al., 2021)) showed perinuclear accumulation (Figure 2B) in 33.3% ± 6.0% and 50% ± 11.6% of infected cells, respectively (Figure 2C). ACAP1 and ACAP2, which act as GAPs for ARF6 downstream of Rab10 (Shi and Grant, 2013) accumulated perinuclearly (Figure 2B) in a similar fraction of cells as Rab10 (58.1% ± 7.1% and 61.1% ± 12.9% of infected cells, respectively; Figure 2C). Unfortunately, we were unable to accurately monitor DENND4, the Rab5-dependent GEF for Rab10 (Yoshimura et al., 2010), and MICAL-L1, the Rab10 effector associated with tubulation (Farmer et al., 2021), with the available antibody reagents. Essentially identical results were obtained on Balb 3T3 cells at 16 hpi, and some of these Rab10 effectors accumulated to a similar extent as Rab10 at six hpi (Supplementary Figure S6). Colocalization analysis of endogenous EHBP1 and Rabin8 was performed on NIH 3T3 GFP-Rab10 cells after 24 h induction of EGFP-Rab10 expression, as all antibody reagents were of rabbit and simultaneous staining with endogenous Rab10 was not possible. As expected, perinuclearly accumulated Rab10 colocalized with EHBP1 and Rabin8 (Supplementary Figure S7). These data show perinuclear accumulation of Rab10 interactors supporting the conclusion of expansion of Rab10-PD in the area of inner pre-AC.
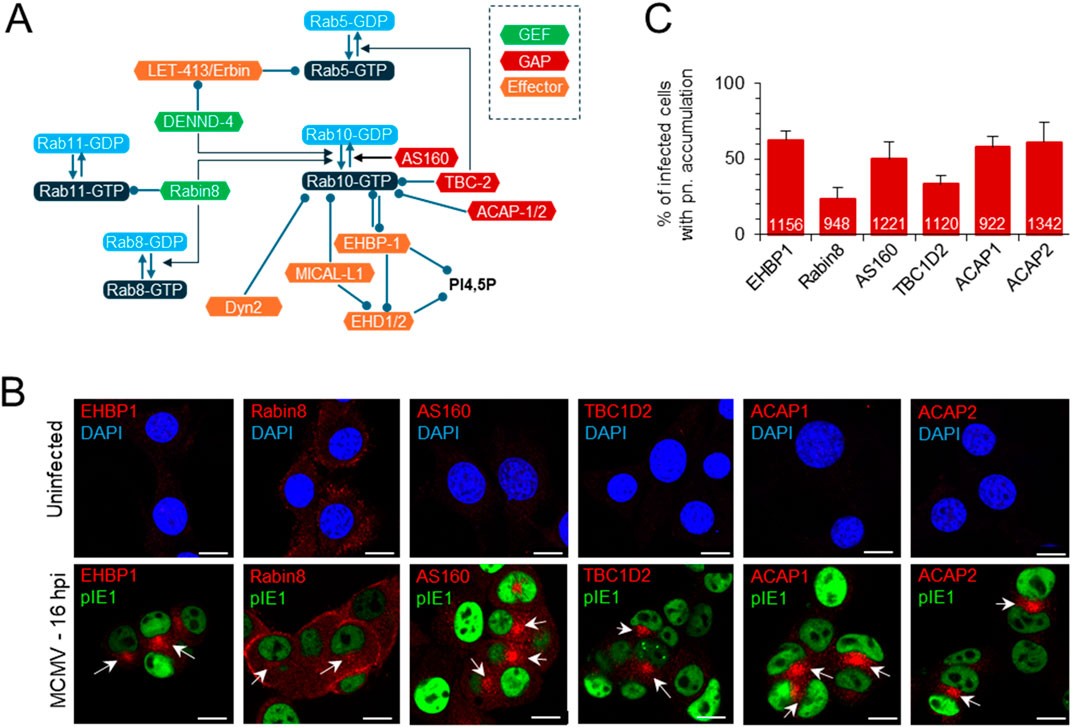
Figure 2. Perinuclear accumulation of EHBP1, Rabin8 and Rab10 effectors within the pre-AC. (A) Schematic representation of the regulatory network involving Rab10 at the EE-RE/ERC interface, Rab10 interactors and effectors. Arrows indicate activity and circles recruitment. GEF, guanine nucleotide exchange factors; GAP, GTPase-activating proteins. (B) Immunofluorescence analysis of EHBP1, Rabin8 and Rab10 effectors in uninfected and MCMV-infected cells. NIH 3T3 cells were infected or uninfected with Δm138-MCMV (MOI of 10) and fixed at 16 hpi, permeabilized and stained with Abs against EHBP1, Rabin8, AS160, TBC1D2, ACAP1 or ACAP2 (red) in combination with Abs against pIE1 in infected cells to control infection (green) or with DAPI (blue) in uninfected cells to stain nuclei. Shown are the merged confocal images through the focal plane of a representative experiment. The arrows indicate pericentriolar accumulation in the pre-AC. Bars, 10 μm. (C) Percentage of cells with perinuclear (pn) accumulation in MCMV-infected (pIE1-positive) cells, shown as mean ± SD from three independent experiments. The numbers in the bars indicate the total number of cells analyzed.
The accumulation of Rab10 and its known interactors at EE-RE/ERC is not associated with increased transcriptional activity (Supplementary Figure S8), as shown by the analysis of our previously published transcriptome (Lučin et al., 2020) and total protein accumulation, as shown by Western blot analysis of Rab10, EHBP1, and Rabin8 in Balb 3T3 cells (Supplementary Figure S6C). Thus, the expansion of Rab10-PD is associated with prolonged retention of Rab10 and its interactors at membranes within the inner pre-AC.
3.3 Rab10 interacts with EHBP1 and MICAL-L1 in MCMV infected cellsThe accumulation of Rab10 and its interactors at the membranes of the inner pre-AC suggests Rab10-based recruitment and retention and should be detectable by a biochemical method to identify protein interactions. To test this hypothesis, we used the proximity-dependent biotin identification (BioID) assay (Roux et al., 2012; Sakai et al., 2019) to identify proteins in close proximity (10–20 nm) to Rab10. We fused Rab10 with BioID2 and HA and generated an NIH 3T3 Rab10-BioID2-HA cell line expressing Rab10-BioID2-HA fusion protein with an expected molecular weight of 54.2 kDa (Figures 3A, B). In this cell line, Rab10-BioID2-HA was visualized in the cytosol in membrane structures that exhibited vesicular and tubular patterns (Figures 3C, D). After MCMV infection, Rab10-BioID2-HA concentrated in the perinuclear region with similar kinetics as in paternal NIH 3T3 cells, as shown by immunofluorescence visualization with anti-Rab10 and anti-HA antibodies (Figures 3C, D). To investigate whether the biotin ligation response occurs at the sites of Rab10 accumulation, we exposed MCMV-infected cells to biotin for 18 h during the early phase of infection. Staining with streptavidin-Alexa-Fluor 488 (SA-AF488) revealed an accumulation of biotinylated proteins in the pre-AC that strongly overlapped with areas expressing Rab10-BioID2-HA, as shown by staining with anti-HA antibodies (Figure 3E; Supplementary Figure S9).
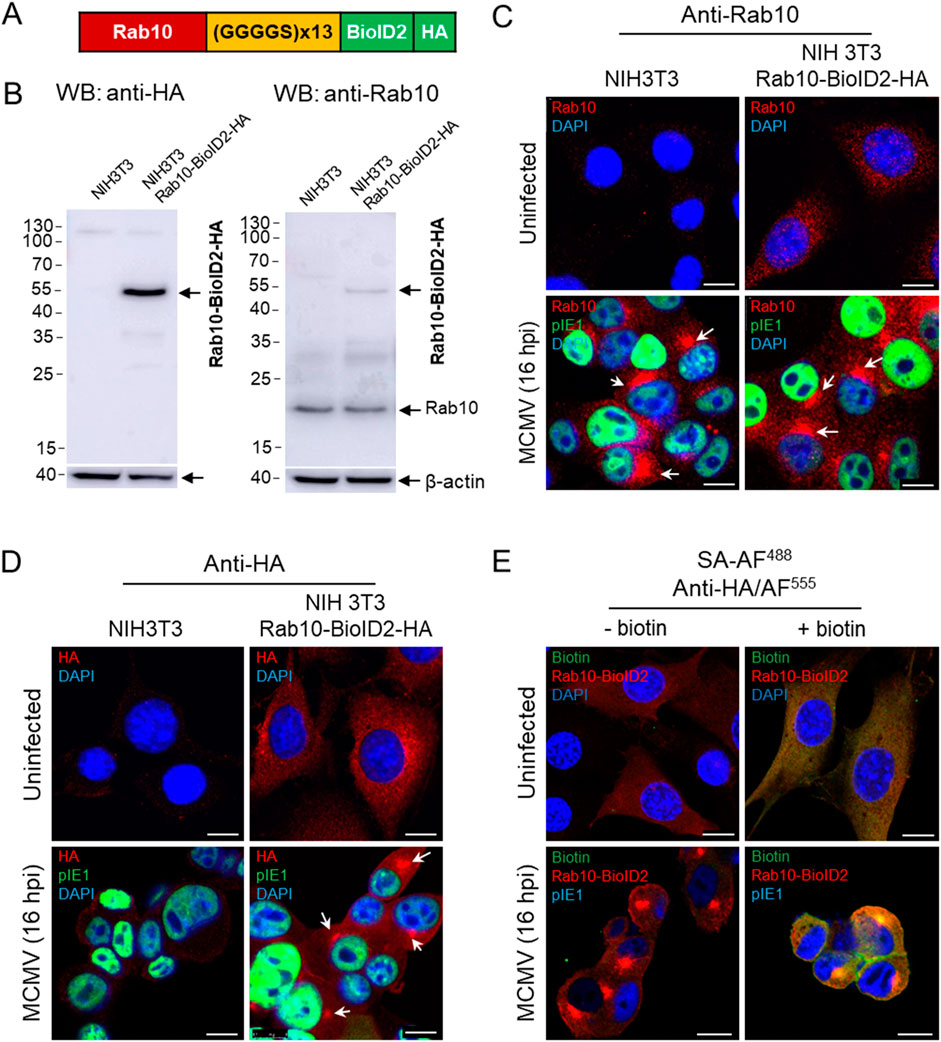
Figure 3. Establishment of the NIH 3T3 Rab10-BioID2-HA cell line. The Rab10-(GGGGS)13-mBioID2-HA fusion sequence (A) subcloned into the pGenLenti lentiviral vector was used to establish the NIH 3T3 Rab10-BioID2-HA cell line, which was analyzed by Western blot (B) for the expression of Rab10-BioID2-HA using antibodies against HA and Rab10. (C, D) Immunofluorescence analysis of uninfected and MCMV-infected (Δm138-MCMV at an MOI of 10 for 16 h) paternal and NIH 3T3 Rab10-BioID2-HA cells stained with antibodies against Rab10 and HA (red fluorescence). DAPI was used to stain the nucleus and anti-pIE1 (green fluorescence) to visualize the infected cells. Shown are the merged confocal images through the focal plane of a representative experiment. Arrows show the perinuclear accumulation of Rab10 and Rab10-BioID2-HA in paternal and NIH 3T3 Rab10-BioID2-HA. (E) Immunofluorescence analysis of the biotinylation reaction in NIH 3T3 Rab10-BioID2-HA cells treated with biotin. Uninfected and MCMV-infected (Δm138-MCMV at an MOI of 10) cells were treated with biotin (50 μM) and after 18 h fixed, permeabilized and stained with AF488-conjugated streptavidin (SA), anti-HA and anti-IE1, followed by AF555- and AF680-conjugated non-cross-reactive secondary antibodies, respectively. The complete set of images is shown in Supplementary Figure S9. Bars, 10 μm.
To identify proteins that interact with Rab10, we exposed Rab10-BioID2-HA-expressing cells to biotin for 18 h (Figure 4A). Cells were uninfected (Ø), infected together with the addition of biotin, or infected after 12 h in the biotin-containing medium. After 18 h, uninfected, six hpi, and 18 hpi samples were lysed and the lysates were precipitated with NeutrAvidin (NA) agarose to pull-down (PD) biotinylated proteins and analyzed by Western blot. To visualize the proteins that were in close contact with Rab10-BioID2-HA, the membranes were stained with antibodies against Rab10 (Figure 4B), the Rab10 interactors (Figure 4C), and three control proteins that are functionally related to Rab10 at the EE-RE/ERC interface but are not expected to establish close contact with Rab10 (Figure 4D). The whole cell lysates (WCL) served as a control to show the endogenous levels of these proteins (Figures 4B–D). Detection of MCMV protein pIE1 and β-actin served to identify the level of infection and loading control, respectively, at the same membrane as biotinylated host cell proteins (Figures 4B–D).
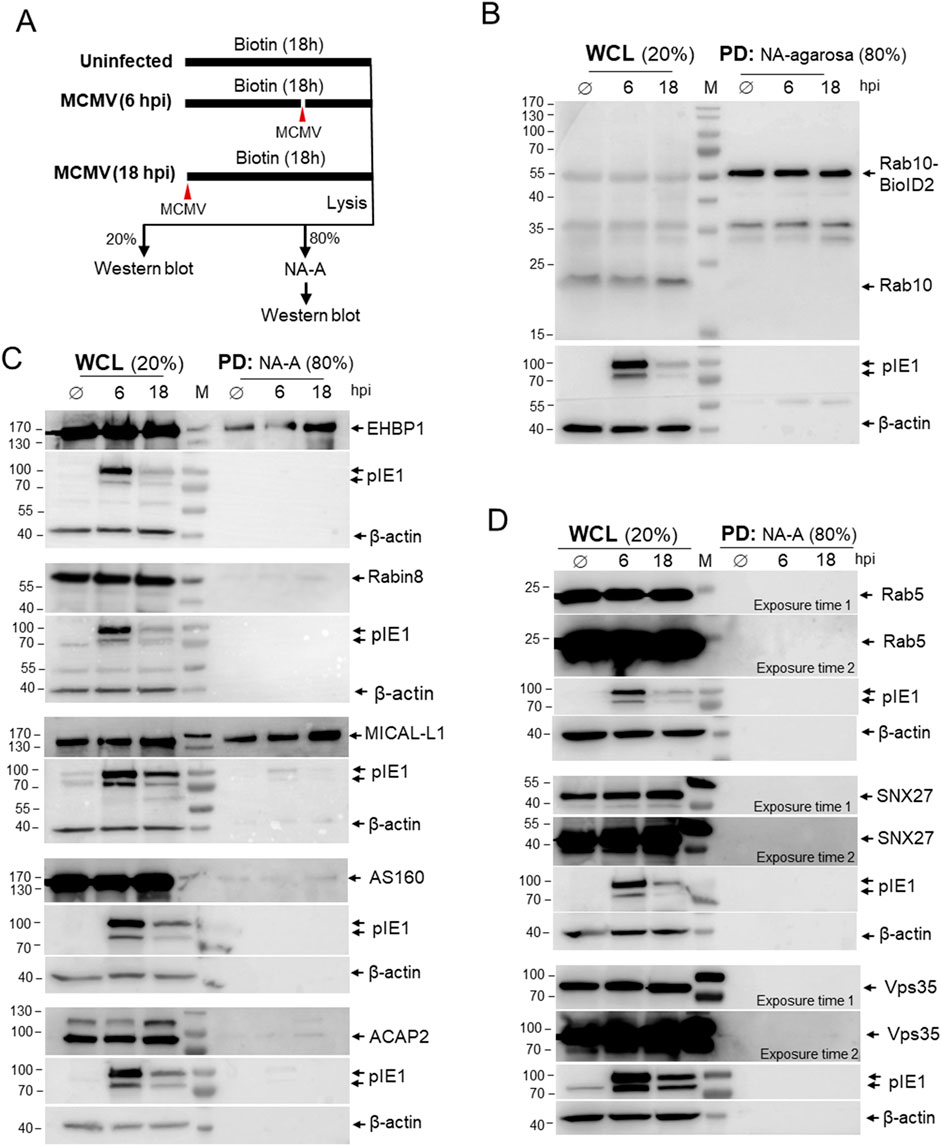
Figure 4. Proximity-dependent biotin identification (BioID) of Rab10 interactors in MCMV-infected cells. (A) Schematic representation of the BioID assay. NIH 3T3 Rab10-BioID2-HA cells were exposed to biotin and left uninfected (Ø) for 18 h or infected with Δm138-MCMV (MOI of 10) at one time point or 12 h after biotin and incubated in biotin for a total of 18 h to reach 18 and 6 h post-infection, respectively. Cell samples were lysed and 20% of the sample was used for Western analysis as whole cell lysate (WCL) and 80% of the sample was used for pull-down (PD) with NeutraAvidin-Agarose (NA-A) and subsequent Western blot analysis. The membranes with WCL and NA-A pull-down (PD) samples were analyzed with (B) antibodies against Rab10 and (C) antibodies against Rab10 interactors: EHBP1, Rabin8, MICAL-L1, AS160 and ACAP2; and (D) non-interacting cellular proteins Rab5, SNX27 and Vps35. Expression of pIE1 and β-actin in each sample served as infection and loading controls, respectively, and was performed on the same membranes. Shown are representative blots from four (EHBP1 and MICAL-L1) and two (Rabin8, AS160, ACAP2, Rab5, SNX27 and Vps35) experiments. Original raw blots and unedited ECL images are shown in supplementary (Supplementary Figure S10).
As expected, precipitation with NA agarose (PD) showed that biotinylated Rab10-BioID2-HA were expressed at similar levels in uninfected and MCMV-infected cells at six hpi and 18 hpi (Figure 4B). Although Rab5, SNX27, and Vps35 are recruited to the perinuclear region and are functionally linked to Rab10 accumulation (Lučin et al., 2020; Štimac et al., 2024), these proteins were not biotinylated in MCMV-infected Rab10-BioID2-HA-expressing cells (Figure 4D), suggesting that Rab10-PD does not involve interaction with these proteins and that there is no biotinylation after cell lysis or during immunoprecipitation. On the other hand, the increasing amounts of EHBP-1 and MICAL-L1 (Figure 4C) were precipitated in both uninfected and MCMV-infected cells at 6 and 18 hpi with NA agarose, indicating that Rab10-BioID2-HA generates similar interactions as in uninfected cells and that these interactions increase as the E phase of infection progresses. Biotinylation of MICAL-L1 and EHBP-1 also indicates that Rab10 was mobilized to TREs, as both MICAL-L1 and EHBP-1 are known TRE effectors (Sharma et al., 2009; Etoh and Fukuda, 2019; Farmer et al., 2021). In contrast, little interaction of Rab10-BioID2-HA with Rabin8 was observed at 16 hpi (Figure 4C), suggesting that very little Rab10 was recruited to TREs downstream of Rab11. In addition, very little interaction with AS160 and ACAP2 was identified (Figure 4C), suggesting that the abundant recruitment of Rab10 to TREs in MCMV-infected cells does not involve recruitment of these downstream Rab10 interactors. Thus, the pull-down detection of sustained interactions of Rab10 with EHBP1 and MICAL-L1 in MCMV-infected cells provides biochemical evidence for the expansion of Rab10-PD at EE-derived tubular endosomes, prior to or independent of the Rab11-dependent pathway of their formation.
3.4 Depletion of EHBP1 but not Rabin8 inhibits expansion of Rab10-PD in the early phase of infectionSince EHBP1 and Rabin8 are known Rab10 interactors required for Rab10 mobilization to membranes (Homma and Fukuda, 2016; Rai et al., 2020; Farmer et al., 2021), we observed the development of perinuclear Rab10 accumulation in MCMV-infected cells after knocking them down with siRNA. Knockdown was verified by Western blot (Figure 5A) and by immunofluorescence detection of endogenous proteins with polyclonal antibodies (Figure 5B) after MCMV infection, as endogenous staining of uninfected cells gave only a weak signal. Depletion of EHBP1 reduced the perinuclear accumulation of EHBP1 and consequently the expansion of Rab10-PD, whereas treatment with scr-RNA and depletion of Rabin8 resulted in perinuclear accumulation of Rab10-PD as in non-transfected cells (Figures 5C, D; Supplementary Table S1). The inhibitory effect of EHBP1 depletion was also observed in Balb 3T3 cells (Supplementary Figure S11). These data indicate that EHBP-1, but not Rabin8, is required for Rab10 recruitment and Rab10-PD expansion, supporting our observation that expansion does not occur downstream of Rab11-mediated recruitment of Rabin eight to the ERC, at membranes of Rab11-REs (Homma and Fukuda, 2016).
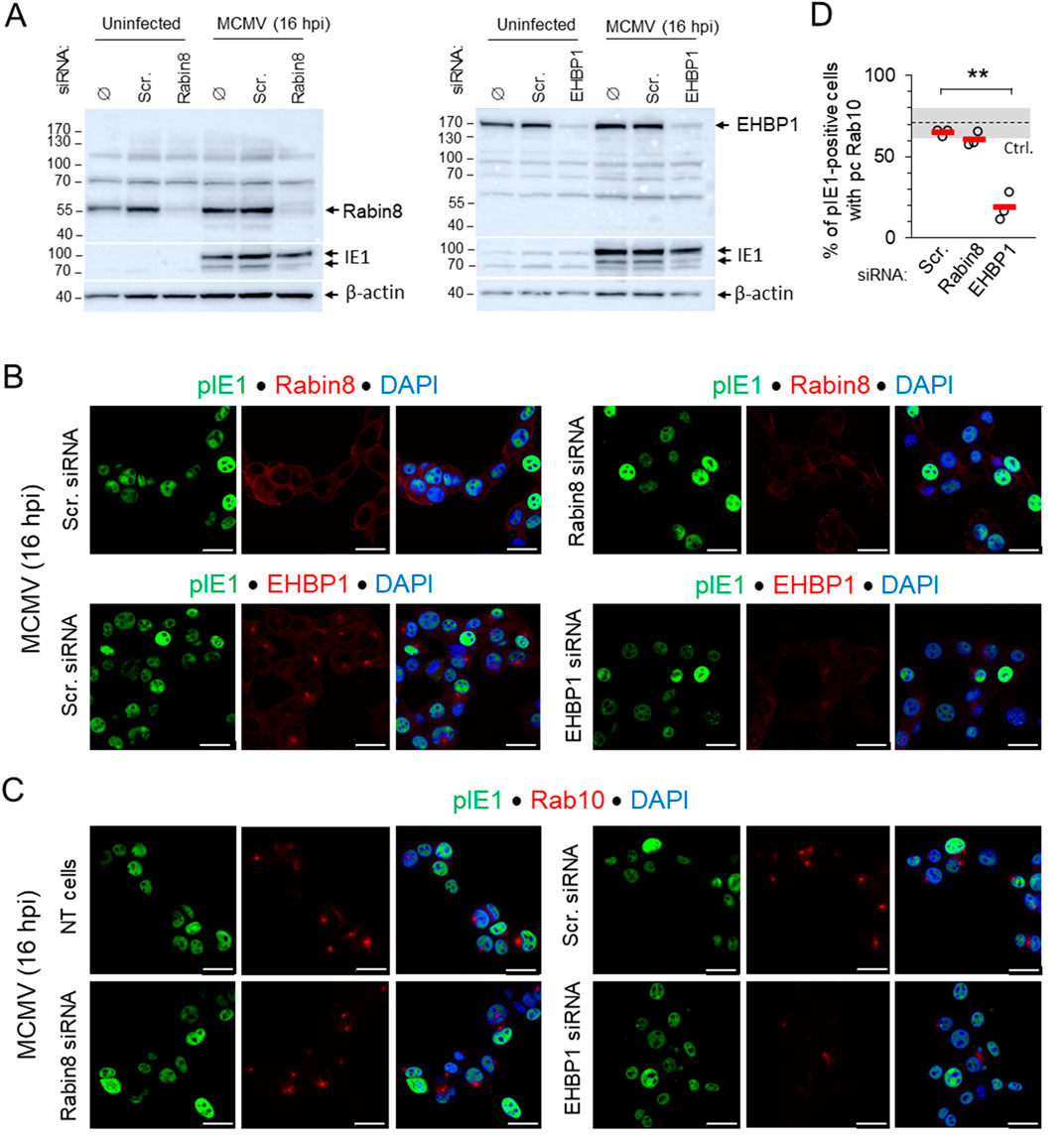
Figure 5. Depletion of EHBP1, but not Rabin8, prevents perinuclear expansion of Rab10-PD in pre-AC. (A) NIH 3T3 cells were transfected with siRNA for EHBP1 or Rabin8. Control cells were either not transfected (Ø), or transfected with “scrambled” siRNA. After 48 h, cells were infected with Δm138-MCMV (MOI of 10) for 16 h, or left uninfected, and lysed for Western blot analysis. Expression of EHBP1 and Rabin8 was determined on the same membrane along with pIE1 as a control for infection and β-actin as a loading control. (B) Immunofluorescence detection of endogenous EHBP1 and Rabin8 (red) in Scr. siRNA- and EHBP- or Rabin8-siRNA-treated cells at 16 h post-infection. Staining for pIE1 (green) served as a control for infection and DAPI for visualization of nuclei. Shown are the confocal images through the focal plane. (C) Immunofluorescence detection of endogenous Rab10 (red) in untreated (NT), Scr. siRNA- and EHBP1 or Rabin8 siRNA-treated cells. Staining for pIE1 (green) served as a control for infection and DAPI for visualization of nuclei. Shown are the confocal images through the focal plane. Bars, 25 μm. (D) Percentage of MCMV-infected (pIE1-positive) cells with perinuclear accumulation of Rab10 in Scr. siRNA- and EHBP1 or Rabin8 siRNA-treated cells 16 h post-infection, shown as mean ± SD. The total number of cells analysed: Scr 1,659; EHBP1 1,578; Rabin8 1,578. Ctrl., control level. Statistical significance was determined by Student’s t-test (**p < 0.01).
3.5 Saturation of PI(4,5)P2 inhibits expansion of Rab10/EHBP1-positive domains within the pre-ACRab10 generates and maintains EE-derived tubular recycling endosomes (TREs) by binding to EHBP1 which is recruited to PI(4,5)P2-enriched membrane domains (Etoh and Fukuda, 2019; Farmer et al., 2021) (Figure 6A). To test whether perinuclear expansion of Rab10-PD requires the establishment of PI(4,5)P2 microdomains on membranes, we transfected cells with EGFP-PH-PLC-δ1 constructs that bind and competitively saturate PI(4,5)P2 on membranes (Tan et al., 2015). In uninfected cells, this construct mainly labels PM and peripheral membranes, a major site of PI(4,5)P2 accumulation (Tan et al., 2015), but also perinuclear membranes, possibly comprising tubular endosomal structures downstream of PI3P-rich EE domains (Figure 6B; Supplementary Figure S12A). In MCMV-infected NIH 3T3 cells, expression of EGFP-PH-PLC-δ1 was accompanied by a reduction in perinuclear accumulation of EHBP-1 and Rab10 that was inversely proportional to the level of EGFP-PH-PLC-δ1 expression (Figures 6C, D; Supplementary Table S2). The same was also observed in Balb 3T3 cells (Supplementary Figures S12B, 12C; Supplementary Table S3). These data confirm that the expansion of Rab10/EHBP1-PD in the inner pre-AC of MCMV-infected cells is associated with PI(4,5)P2 membrane microdomains downstream of the PI3P-rich membrane domains of EEs, which most likely resemble TREs.
留言 (0)