
記住我
The earliest experimental evidence supporting the presence of malignancy stem cells (MSCs) in humans was reported in 1994 (Lapidot et al., 1994) and has since been the subject of extensive research, contributing to a better understanding of tumorigenesis and the development of more effective anti-malignancy therapies (Clarke and Fuller, 2006; Fulawka et al., 2014; Yadav and Desai, 2019; Lica et al., 2024; Zhang et al., 2024). Stem cells (SCs) are undifferentiated, primitive cells in an organism (Fuchs and Segre, 2000; Weissman, 2000; Bongso and Lee, 2005; Brignier and Gewirtz, 2010). When cultured in vitro, these cells often undergo epigenetic changes and exhibit dysfunctions; therefore, they are better described as stem-like cells (SLCs) (Nestor et al., 2015; Weissbein et al., 2017; Bar and Benvenisty, 2019; Liu et al., 2020; Tognon et al., 2021; Kabacik et al., 2022).
The initial evidence suggesting the existence of SCs in the human body can be traced back to a study published in 1959, which demonstrated that transplanted bone marrow cells could regenerate the hematopoietic system in patients who had undergone intensive whole-body irradiation, indicating the existence of hematopoietic SCs (HSCs) (Thomas et al., 1959). The first detailed description of healthy HSCs derived from long-term in vitro cultures was published in 1980, where they were referred to as long-term culture-initiating cells (Gartner and Kaplan, 1980). Today, they would more accurately be called hematopoietic SLCs (HSLCs).
In cancer research, a subset of SCs has also been identified. Malignant cells (MCs) originating from epithelial tissues are known as cancer SCs (CSCs), while those from other germ layers have specific names; for example, in leukemia, they are termed leukemic SCs (LSCs) (Lapidot et al., 1994). The CSC hypothesis was initially considered controversial (Bhagwandin and Shay, 2009; Magee et al., 2012) but has gained broad acceptance over time (Batlle and Clevers, 2017; Phi et al., 2018; Najafi et al., 2019; Saygin et al., 2019; Liu et al., 2023). Outside the organism, MSCs, including CSCs and LSCs, are called cancer SLCs (CSLCs) (Kondo et al., 2004) or leukemic SLCs (LSLCs) (Bonnet and Dick, 1997).
Evidence for the presence of LSLCs in cell lines emerged in the 1990s and 2000s, primarily in studies focused on leukemia cell lines such as HL60 (Bonnet and Dick, 1997). The stage-specific heterogeneity of this and other acute myeloid leukemia (AML) cell lines enabled the identification of a small subpopulation of LSLCs. These cells exhibited symmetric division (SD) capabilities, the ability to initiate and drive disease progression, and cytological and morphological similarities to LSCs (Lapidot et al., 1994; Bonnet and Dick, 1997; Guan et al., 2003). Notably, the HL60 cell line was established in the 1970s through the leukapheresis of peripheral blood from a patient with AML, and even at that time, the presence of cells with LSLC properties capable of initiating AML in NOD mice was suggested (Collins et al., 1977; Gallagher et al., 1979). The first HL-60 leukopoiesis model was proposed in 1988, identifying LSLCs at the top of the hierarchical development of this cell line (Birnie, 1988), which appears to support subsequent mathematical models of hematopoietic senescence (Marciniak-Czochra et al., 2009; Stiehl et al., 2014).
Following the discovery of LSLCs, CSLCs were identified in cultures from solid tumors, including those from brain and breast cancers (Ignatova et al., 2002; Al-Hajj et al., 2003; Singh et al., 2004). Glioblastoma CSCs (U87) were discovered in 2002, and they had been characterized using the PROM1 marker (formerly known as cluster of differentiation 133 - CD133) by 2004 (Ignatova et al., 2002; Singh et al., 2004). These cells demonstrated the capacity to SD, differentiate into various cell types, and initiate tumor growth in vivo when transplanted into animals, exhibiting traits similar to neural SCs. In 2003, CSLCs were isolated from the MCF7 breast cancer cell line and identified by the surface markers CD44⁺/CD24⁻ (Al-Hajj et al., 2003). These cells exhibited a tumor-initiating capacity in immunodeficient mice and shared key CSC features, such as SD and the ability to differentiate into multiple lineages.
Unlike multipotent SCs such as HSCs, pluripotent SCs (PLSCs) can develop into all cell types in the body, distinguishing them from tissue-specific SCs such as HSCs. Human PLSCs from an embryo, called embryonic stem cells (ESCs), were first successfully isolated and described in 1998 (Thomson et al., 1998). Shortly thereafter, in vitro cultures containing equivalent cells, termed pluripotent-like SCs and embryonic-like SCs (ELSCs), were established (Amit et al., 2000). In 2007, induced PLSCs (iPLSCs) were created from adult human fibroblasts through reprogramming with key transcription factors (octamer-binding transcription factor 4 [OCT4], SRY-box transcription factor 2 [SOX2], Kruppel-like factor 4 [KLF4], and MYC protooncogene [c-MYC]). These iPLSCs behave like ESCs and can differentiate into various cell types (Takahashi et al., 2007).
One of the most primitive ESCs in the human body are cells derived from the inner cell mass (ICM) of the blastocyst. Epiblast cells (EpibCs) form from the ICM in the pre-gastrulation stage and play a key role in early embryo development (Tam and Loebel, 2007; Rossant, 2008). EpibCs are the source of the germ layers (ectoderm, mesoderm, and endoderm), as well as extraembryonic cells, which forms structures supporting embryo development, including the chorion (a part of the placenta) and the allantois (Arnold and Robertson, 2009; Hayashi and Saitou, 2013; Saitou and Miyauchi, 2016).
The EpibCs also generate primordial germ cells (PGCs), which in humans appear around the second to third week of embryonic development, originating from the epiblast. PGCs migrate from the posterior part of the embryo to the developing gonads (testes or ovaries), where they later differentiate into gametes. Studies on human embryos have shown that PGCs emerge early in development and are crucial for forming the germline. These cells play vital roles in embryo development and the formation of supporting structures, enabling proper fetal development (Rossant and Tam, 2017).
During gastrulation, EpibCs migrate into the embryo, giving rise to ectodermal progenitor cells (EcPCs), mesoderm progenitor cells (MdPCs), endodermal progenitor cells (EnPCs), and PGCs (Tam and Behringer, 1997). EcPCs give rise to neural precursor cells, which differentiate into various nervous system cells, including neurons, astrocytes, and oligodendrocytes, as well as epidermal precursor cells, which form skin, epidermal, and glandular cells. EnPCs are responsible for forming the digestive and respiratory systems, including the liver, pancreas, and lungs, through pathways such as hepatic and pancreatic progenitor cells. MdPCs develop into progenitor and precursor cells: hematopoietic precursor cells (HPCs), which will give rise to blood and lymphatic cells; mesenchymal precursor cells (MsPCs), responsible for forming muscle, bone, cartilage, and adipose tissue; cardiac precursor cells, which generate various heart cells; and endothelial progenitor cells (EnthPCs), involved in blood vessel formation (Figure 1).
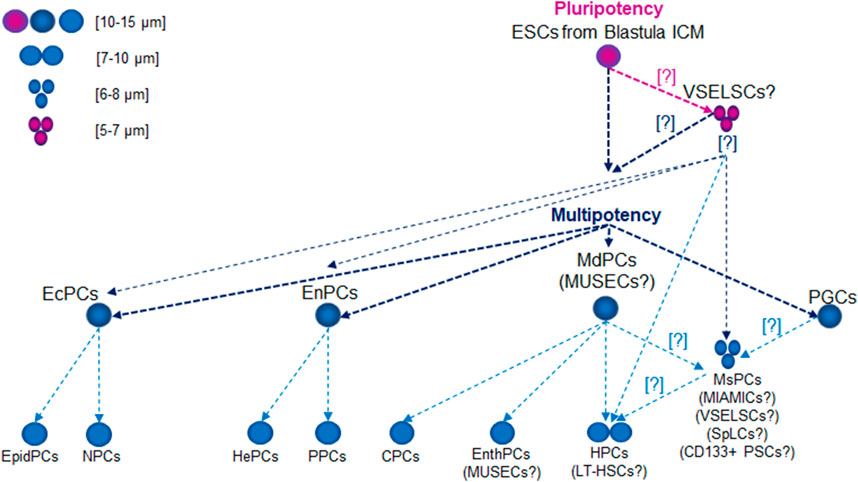
Figure 1. Lifetime Development of Primitive Stem Cell Precursors and Progenitors. Abbreviations: CPCs, cardiac precursor cells; CD133+ PSCs, CD133+ precursor cells; EcPCs, ectodermal progenitor cells; EnthPC, endothelial progenitor cells; EnPCs, endodermal progenitor cells; EpidPCs, epidermal precursor cells; ESCs, embryonic stem cells; HePCs, hepatic precursor cells; HPC, hematopoietic precursor cells; ICM, inner cell mass; LT-HSCs, long-term hematopoietic stem cells; MdPCs, mesoderm progenitor cells; MIAMICs, marrow-isolated adult multilineage inducible cells; MsPCs, endothelial progenitor cells; MUSECs, multilineage-differentiating stress enduring cells; NPCs, neural precursor cells; PGCs, primordial germ cells; PPCs, pancreatic progenitor cells; SpLCs, spore-like cells; VSELSCs, very small embryonic-like stem cells.
Embryonic cancer cells (ECCs), also called embryonal carcinoma cells, are cancer cells that originate from embryonic tumors, particularly those related to germ cell tumors (germinomas) or other cancers developing from PGCs, such as embryonal carcinoma. ECCs were first mentioned in research on germ cell tumors, particularly mouse embryonal carcinoma (Stevens, 1958). These cells were identified and described in teratocarcinoma: tumors derived from pluripotent cells that can differentiate into various tissue types. Further studies on teratocarcinoma revealed that ECCs represent the malignant counterpart of pluripotent cells, maintaining the ability to SD and differentiate like normal ECs, but their growth is uncontrolled (Pierce et al., 1959). Early research on ECCs focused on tumors linked to the gonads (Damjanov and Solter, 1974). These ECCs arise from PGCs, which typically develop into gametes like sperm or eggs. When these cells undergo abnormal development or mutations, they can cause cancer, forming germ cell tumors, including embryonal carcinomas (Moraru et al., 2023). These tumors are mostly found in the testes (testicular cancer) or ovaries but can also occur in other regions, such as the mediastinum or brain, as extragonadal germ cell tumors like teratomas (Miller et al., 2014; Zhu et al., 2018; Kidder, 2024). ECCs are pluripotent, meaning they can differentiate into many cell types, similar to normal SCs (Cunningham et al., 2012; Gropp et al., 2012; Montilla-Rojo et al., 2023). However, unlike healthy SCs, ECCs grow uncontrollably, leading to cancer development. ECCs contribute significantly to the composition of heterogeneous tumors, such as teratomas and mixed embryonal carcinomas, which contain various tissues.
The precursor stages of MSCs have garnered significant interest, particularly in exploring the mechanisms of carcinogenesis (Bhartiya et al., 2023b). Recent experimental evidence indicates that CSCs can originate from malignantly transformed very small ELSCs (VSELSCs) (Bhartiya et al., 2023b).
The existence of CSCs from transformed VSELSCs in vivo has been exemplified by very small CSLCs (VSCSLCs) and LSLCs (VSLSLCs) observed in vitro (Lica and Pradhan, 2023). Notably, VSELSCs were discovered in the human body in 2007 (Kucia et al., 2007) and, like CSCs and SCs, were initially met with skepticism. However, numerous independent laboratories have since confirmed their existence in vivo (Ratajczak et al., 2019), and recent reports about their cell culture counterparts have generated considerable interest (Lica and Pradhan, 2023). The latter has opened up avenues for investigating leukemic and cancer SCs precursors in vitro, given the availability of experimental material in the form of cell cultures and the potential for efficient methods to enrich the number of primitive stages (Lica et al., 2018, 2021; Lica and Pradhan, 2023). There is a promising opportunity to compare the most primitive stages of healthy cells and MCs by obtaining a minimum of 5,000 (preferably 10,000) cell stages for single-cell RNA sequencing (scRNA-seq) analysis (Danielski, 2023). These results increase our chances of understanding the molecular mechanisms underlying malignant proliferation, as well as differences in cell cycle checkpoints (Skladanowski et al., 2009) and the types of cell stage division according to the Hayflick effect (HE) (Hayflick, 1965) between healthy VSELSCs and transformed VSCSCs as well as VSLSCs, along with their stage in vitro analogs. The abbreviations currently used and suggested for very small SC progenitors and precursors in vivo and in vitro are presented in Table 1.
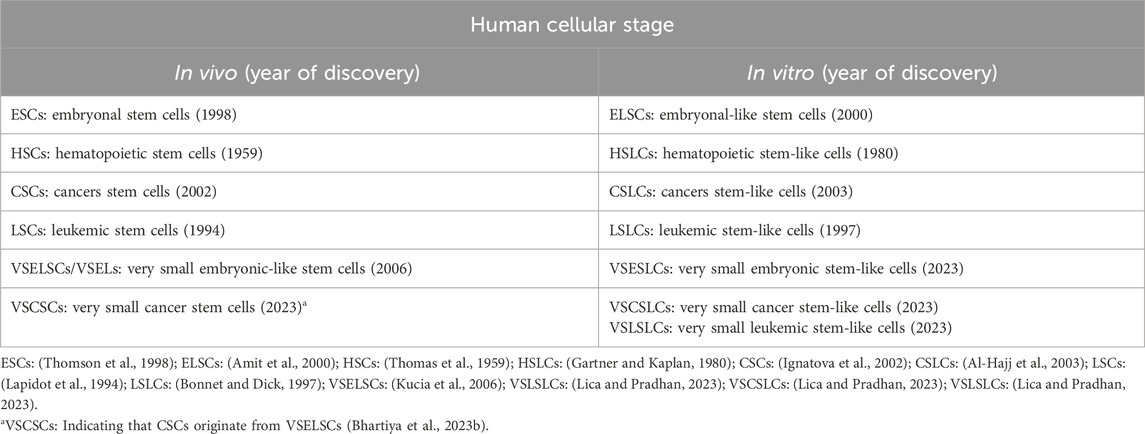
Table 1. Very primitive cellular stages in vitro and in vivo.
Origin of VSELSCsIndependent studies have detected biological material morphologically typical of VSELSCs in human tissues across various age groups, from young to elderly individuals (Virant-Klun et al., 2008; Sovalat et al., 2011, 2016; Chang et al., 2014; Vojnits et al., 2014; Kakavoulia, 2021). In suitable experimental models, VSELSCs have exhibited pluripotent or multipotent capabilities (Bhartiya et al., 2012; Vojnits et al., 2014; Bhartiya, 2015), differentiating into various cell types, including skeletal cells (Havens et al., 2012), vascular endothelial cells, cardiomyocytes (Wu et al., 2011), lung epithelial cells (Jin et al., 2013), and male or female gametes (Virant-Klun et al., 2008, 2013; Bhartiya et al., 2012; Bhartiya, 2015).
However, critics argue that VSELSCs lack pluripotent characteristics (Danova-Alt et al., 2012; Ivanovic, 2012; Alvarez-Gonzalez et al., 2013; Szade et al., 2013). Supporters of VSELSCs contend that discrepant findings may arise from differences in research protocols.
They also emphasize that VSELSCs share molecular similarities with early-stage migratory PGCs, which, upon restoration of somatic genomic imprinting, resemble EpibCs capable of proliferating in vitro and differentiating into all three germ layers (Shin et al., 2010). It is hypothesized that VSELSCs may originate from PGCs, derived from the EpibCs during the early stages of embryogenesis, typically within the first weeks of human development (Kucia M. et al., 2008; Ratajczak et al., 2013b; Ratajczak et al., 2019; Barati et al., 2021; Bhartiya et al., 2022; Bhartiya et al., 2023a). PGCs migrate to the developing gonads, where they differentiate into spermatogonia (sperm precursors) in males or oogonia (egg precursors) in females. When isolated and cultured under specific conditions, primordial germ-like cells (PGLCs) can be reprogrammed into pluripotent embryonic germ-like cells (EGLCs), which can differentiate into various cell types, like ESCs, albeit with epigenetic differences (Durcova-Hills et al., 2008; Panula et al., 2010; Nagamatsu et al., 2013; Kurek et al., 2020; Tran et al., 2021; Jo et al., 2022; Reda et al., 2022). The size of human ESLCs and PGLCs depends on their developmental stage and culture conditions. ESLCs, with an average size of 10–15 μm, are small with large nuclei due to their high metabolic activity and rapid division. PGLCs, slightly larger at 12–20 μm, exhibit size variations influenced by culture conditions and developmental cues, reflecting their preparation for migration and differentiation observed in early embryogenesis.
Recent research has demonstrated methods for differentiating human SCs into PGLCs, which mimic the behavior of PGCs during embryogenesis, particularly when cultured with specific signals such as bone morphogenetic protein 4 (BMP4) and Nodal protein (NODAL) (Shamblott et al., 1998). This differentiation allows the early steps of human germ cell development to be studied even though these cells do not exist naturally in adult human bodies. PGLCs have a limited capacity for directly forming tissue-specific SLCs; however, their pluripotent descendants, such as EGLCs, can differentiate into various tissue types in vitro (Yu et al., 2007; Durcova-Hills et al., 2008; Panula et al., 2010; Nichols and Smith, 2012; Nagamatsu et al., 2013; Katajisto et al., 2015). The observed pluripotency of cells exhibiting the cytological features of VSELSCs indicates that their proliferative potential exceeds that of PGCs.
The asymmetric division (AD) of PGCs has been well-studied in mice, although research on humans is more limited (Rossant and Tam, 2009; Bedzhov and Zernicka-Goetz, 2014). Much of what is known about PGC mechanisms comes from indirect observations and in vitro studies of PLGCs. Studies have shown that these cells undergo ADs, leading to gamete differentiation (Surani et al., 2007; Perrett et al., 2008; Gkountela et al., 2013; Tang et al., 2016). In humans, this suggests that PLGC division results in one retained SC and another differentiating cell (Tang et al., 2016). The retained SC continues to divide and differentiate into germ cells, while the daughter cell begins differentiating into more mature forms of germ cells, such as oogonia in females or spermatogonia in males. The ADs of PGCs and PLGCs ensure the SC pool is maintained while allowing other cells to differentiate into more advanced developmental stages (Gkountela et al., 2013; Tang et al., 2016). Research on SC dynamics has explored how the balance between SD and AD can fluctuate based on environmental cues and developmental stages. SD may be activated to replenish lost SCs during injury or periods of rapid growth, while maintaining homeostasis often requires AD to prevent uncontrolled cell growth (Shahriyari and Komarova, 2013). Disruptions in AD have been linked to cancerous growth due to unchecked cell division (Hansen and Pelegri, 2021). Specifically for PGLCs, studies indicate they can transform into EGLCs, which share characteristics with PLSCs. These EGLCs demonstrate high SD capabilities and can proliferate through SD under specific in vitro conditions (Durcova-Hills et al., 2008). While evidence favors PGCs undergoing AD during development, their capacity for SD in vitro, particularly during reprogramming or under specific growth conditions, suggests they may behave similarly to SCs in some contexts (Durcova-Hills et al., 2008). During embryonic development, PGCs follow differentiation pathways that result in the formation of oocytes or spermatogonia. These processes are tightly regulated and typically occur when PGCs transition from SD to AD, producing differentiated offspring. Reprogramming of PGLCs indicates these cells may possess a previously unrecognized level of plasticity; however, this reprogramming does not involve the simultaneous differentiation into less potent cells during their division (Shamblott et al., 1998; Leitch and Smith, 2013).
Another hypothesis regarding the origin of VSELSCs suggests that they derive from ESCs and serve as precursor cells for tissue progenitors, including PGCs (Ratajczak et al., 2019) (Figure 2). These cells are incorporated into developing tissues as OCT4+ cells. The underlying mechanism involves epigenetic modifications of imprinted genes, such as those at the Insulin-like growth factor 2–H19 imprinted maternally expressed transcript (IGF2–H19) and Potassium voltage-gated channel subfamily Q member 1 (KCNQ1)–Cyclin-dependent kinase inhibitor 1C (CDKN1C, formerly known as p57 or Kip2) loci, which maintain these cell stages in a dormant state within adult tissues. A similar process involving the erasure of genomic imprinting also regulates the dormant state of PGCs (Kucia M. et al., 2008; Ratajczak et al., 2019; Ratajczak et al., 2013b).
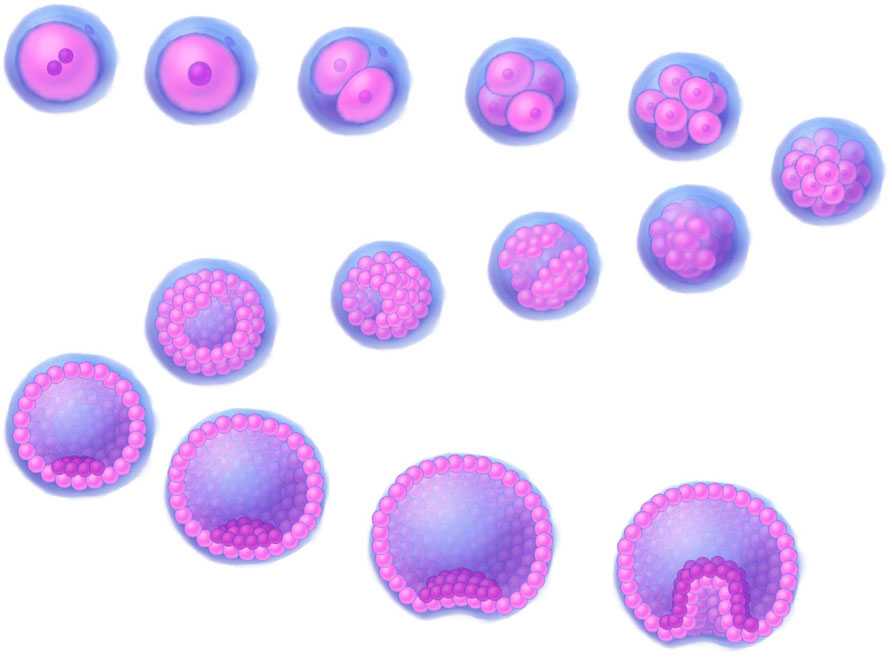
Figure 2. A 3D illustration depicting early stages of human development. Stages of development, progressing downstream from the upper left corner: fertilized egg, zygote, 2-cell stage, 4-cell stage, 8-cell stage, morula (16–32 cells), early blob, late blob, early blastocyst (blastocoel fill <50%), 1_blastocyst (blastocoel fill >50%), 2_blastocyst (blastocoel fill >50%), expanded blastocyst, early gastrula, gastrula. The illustrations were created by graphic artist Mr. Tobiasz Sosnowski, inspired by time-lapse microscopy featured on Iwata et al. (2014) as well on TheDeepSci YouTube channel (accessed 20.11.2024).
Identifying VSELSCs and VSLSLCs: cytological differences from exosomes and cellular debrisIdentifying VSELSCs is particularly complex due to their size and morphological similarities to exosomes, extracellular vesicles (EVs), and apoptotic bodies (ABs) (O'Neill et al., 2012). Despite these challenges, both exosomes and VSELSCs play significant roles in various biological processes (Savina et al., 2003; Doeppner et al., 2015; Machtinger et al., 2015; Yáñez-Mó et al., 2015; Zomer et al., 2015; Tkach and Théry, 2016; Bobis-Wozowicz et al., 2017; Kalluri and LeBleu, 2020). However, their unique ability to SD, grow, and proliferate distinguishes VSELSCs from exosomes and other cellular debris (Kucia M. J. et al., 2008; Ratajczak et al., 2019; Domingues et al., 2022).
EVsEVs are tiny, membrane-enclosed particles released by cells during various biological activities. Their size and function can vary depending on their cellular origin, influencing their environmental interactions and roles in cell-to-cell communication. EVs transport various molecules, including proteins, lipids, nucleic acids, and signaling compounds, through interactions facilitated by their membranes. Importantly, they are not always completely isolated from the external environment by their lipid membranes. Different EV types include ectosomes which protrude from the cell surface and may assist in intercellular signaling; exosomes, which are approximately 30–150 nm in size and can be identified based on specific membrane markers and proteins; microvesicles, which typically measure 0.1–1 µm in size and are released by cells in response to activation, oxidative stress, or inflammation; and apoptosomes, which vary in size and composition based on the stage of apoptosis and cell type and are surrounded by a lipid membrane containing cellular fragments such as nuclear debris or organelles (Kowal et al., 2016; Muhsin-Sharafaldine and McLellan, 2018; Théry et al., 2018; Van Niel et al., 2018; Jeppesen et al., 2019; Crescitelli et al., 2021).
The structural similarities between these EV types and ABs suggest that they could be classified as forms of ABs; conversely, ABs might also be classified as EVs (Table 2). Currently, no specific antibodies target EVs. The presence of specific surface markers can vary depending on the cell type, differentiation stage, physiological processes, microenvironment, and other factors, limiting their specificity. However, some antibodies target surface proteins present on EVs, which can aid their identification and characterization. Common markers and antibodies used in EV studies include tetraspanins (CD9, CD63, CD81), surface markers associated with programmed cell death 6 interacting protein (PDCD6IP) and tumor susceptibility 101 (TSG101), as well as proteins such as heat shock proteins: HSP70, HSP90; and flotillin 1 (FLOT1) (Bobrie et al., 2012; Andreu and Yáñez-Mó, 2014; Li et al., 2017; Merchant et al., 2017; Li et al., 2019; Moeinzadeh et al., 2022; Zhang et al., 2022; Kalluri and McAndrews, 2023).
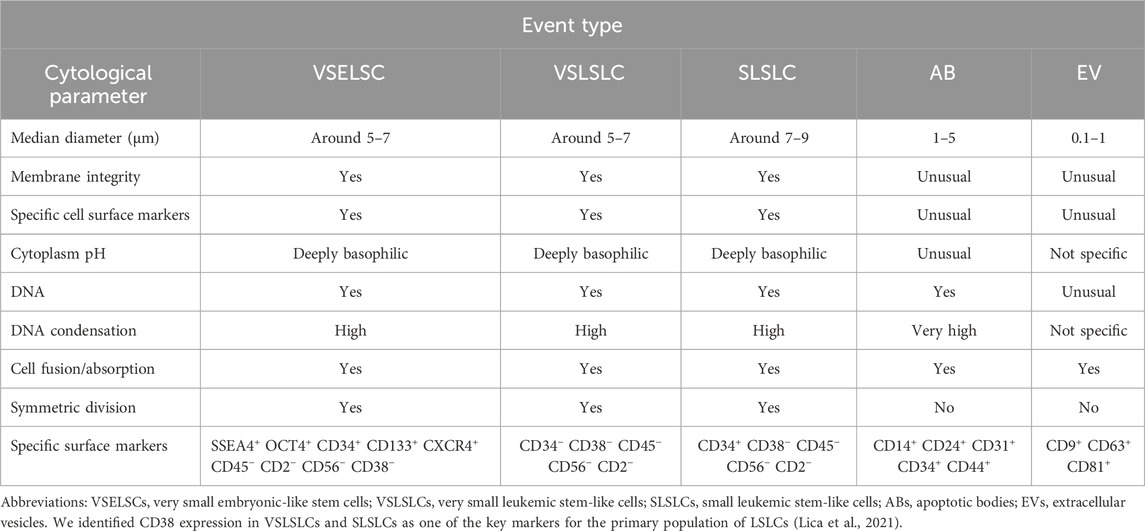
Table 2. Cytological characteristics and comparison of the VSELSCs, VSLSLCs, and SLSLCs with ABs and EVs.
ABsABs are typically small, circular, or oval structures, ranging from 1 to 5 µm in diameter, formed during apoptosis, the programmed cell death process. The cell’s chromatin undergoes significant condensation during apoptosis (Lecoeur et al., 1997; Coleman et al., 2001; Zhang et al., 2018; Aoki et al., 2020; Dou et al., 2020; Serrano-Heras et al., 2020). As the process advances, the cell shrinks, and its fragmented components are encapsulated within vesicles, which may be surrounded by a double membrane, which provides some protection to the contents but does not ensure complete integrity (Kerr et al., 1972; Lecoeur et al., 1997; Coleman et al., 2001; Zembruski et al., 2012; Zhang et al., 2018; Aoki et al., 2020; Dou et al., 2020; Serrano-Heras et al., 2020). Early-stage ABs may not exhibit positive staining for 7-aminoactinomycin D (7-AAD), indicating complete isolation (Montgomery et al., 2020). Importantly, this study did not cover VSELSCs and their morphology. Furthermore, the outer membrane of ABs can display specific CD surface markers, although this is not consistent across all ABs (Kerr et al., 1972; Lecoeur et al., 1997; Coleman et al., 2001; Zhang et al., 2018; Aoki et al., 2020; Dou et al., 2020; Serrano-Heras et al., 2020). The presence of these markers varies depending on the cell type (including the primitive SCs), apoptosis stage, and other factors, making them characteristics with limited specificity.
While exosomes and ABs are distinct entities, they share several features (Table 2). Research on exosomes has often identified common markers such as tetraspanins (e.g., CD63 and CD81), which are also found on ABs (Yáñez-Mó et al., 2015). The complement system is crucial for recognizing and eliminating ABs. The complement protein complex C1q binds to phosphatidylserine and other molecules on AB surfaces, facilitating their clearance by phagocytes and helping to prevent autoimmunity (Païdassi et al., 2008). During apoptosis, DNA and chromatin break apart, exposing histones on the AB surface, which serve as recognition markers (Zernecke et al., 2009). ABs frequently change their membrane protein composition, often losing membrane integrity. Markers associated with ABs include altered membrane proteins such as death receptors (e.g., Fas cell surface death receptor) and apoptosis-regulating proteins from the BCL2 apoptosis regulator family (Nagata, 2018). The tetraspanin CD9 is involved in various cell processes such as adhesion and membrane fusion, and its presence on ABs may enhance cell communication and recognition (Miyanishi et al., 2007). CD14, which binds intercellular adhesion molecule 3 (ICAM3), facilitates interactions between ABs and macrophages for recognition and clearance (Moffatt et al., 1999; Hart et al., 2008). ABs can also express integrins or other adhesion molecules that aid in phagocytic interactions. Exposed on AB surfaces, ICAM3 plays a key role in their recognition by phagocytes (Devitt et al., 2003; Hart et al., 2008). Various lectins that recognize altered sugars on cell membranes and CD14 are important for recognizing and clearing ABs. CD24, associated with cell adhesion and immune modulation, may also facilitate interactions between ABs and immune cells (Liu et al., 2002; Devitt et al., 2003). CD36, a scavenger receptor, promotes AB phagocytosis by recognizing phosphatidylserine on its surface (Greenberg et al., 2006). Platelet endothelial cell adhesion molecule (PECAM1) also known as cluster of differentiation 31 (CD31), another adhesion protein, signals phagocytes when its presence decreases on AB surfaces, marking them for removal (Brown and Savill, 1999). Additionally, the adhesion molecule CD44, which is involved in cell interaction and migration, may contribute to phagocytosis when exposed on ABs (Poon et al., 2014). CD47 typically serves as a “do not eat me” signal for macrophages, but its expression decreases during apoptosis, allowing ABs to be recognized and cleared by the immune system (Gardai et al., 2005). Recent research has highlighted key surface markers on human ABs, particularly phosphatidylserine, that signal phagocytes. Receptors from the TIM and TAM families (hepatitis A virus cellular receptor 1 [HAVCR1], T cell immunoglobulin and mucin domain containing 4 [TIMD4], MER proto-oncogene, tyrosine kinase [MERTK], TYRO3 protein tyrosine kinase [TYRO3], and AXL receptor tyrosine kinase [AXL]) recognize this marker, facilitating the removal of apoptotic cells and regulating inflammation, particularly in cancer and autoimmune diseases (Fadok et al., 2000; Serrano-Heras et al., 2020; Yu et al., 2023; Kur and Weigert, 2024; Lica et al., 2024). Isolating ABs from patient blood samples may offer valuable diagnostic and therapeutic insights, especially for neurological conditions such as stroke and neurodegenerative diseases (Serrano-Heras et al., 2020).
VSELSCsHuman VSELSCs are small cellular structures, typically measuring 5–7 µm in diameter, and exhibit unique embryonic or multipotent characteristics, including the ability to SD and differentiate into various cell types (Sovalat et al., 2011; Shin et al., 2012; Vojnits et al., 2014; Kakavoulia, 2021). While the concept of VSELSCs is promising, further research is needed to elucidate their biological mechanisms and improve the accuracy of detection methods (Abbott, 2013; Nicholls, 2013; Szade et al., 2013; Dulak et al., 2015). Despite these challenges, an increasing number of independent studies support the existence of VSELSCs. Cellular debris, comprising fragments resulting from cellular disintegration, apoptosis, or other cellular activities, can be mistakenly identified as living cells during flow cytometry analysis. Data on global gene expression in murine VSELSCs indicate that key pluripotency markers such as Pou5f1, Nanog, and SRY-box transcription factor 9 [Sox2], along with the involvement of Polycomb proteins, play essential roles in maintaining the pluripotent characteristics of these cell stages (Shin et al., 2012).
VSLSLCsOur earlier studies (Lica et al., 2018, 2021; Lica and Pradhan, 2023) revealed a phenomenon previously observed in healthy human HSCs: the enrichment of culture with primitive stages through cell density control (Csaszar et al., 2012). The LSLCs we identified exhibited an average size of approximately 9–12 µm (Lica et al., 2018, 2021).
Potential VSLSLCs were expected to have a similar size to VSELSCs, LT-LSCs and other small progenitor/precursor SCs (PSCs), which may increase during developmental stage transformations, potentially reaching approximately twice the size during SD or when forming LSLCs (Lica et al., 2018; Lica et al., 2021; Lica and Pradhan, 2023). The corresponding morphological features of these stage transformations should be visible in scatter dot plots, representing the resting phase (around 5–7 µm), SD (up to 10–14 μm, although it might also be 5–7 µm), and AD (up to 13–16 µm), leading to an increase in size through developmental stages. We cannot exclude the occurrence of other ADs at cell sizes of 5–7 μm, which may lead to the formation of smaller, less differentiated cells compared to LSLCs. Given the morphological similarities among hypothetical developmental stages, we have divided the scatter dot plot’s region of interest into two distinct events: VSLSLCs and small LSLCs (SLSLCs). Notably, SLSLCs may represent a developmental stage of VSLSLCs undergoing by SD or AD changes in DNA distribution. Identifying specific CD markers that uniquely target SCs, CSCs, LSCs, and their precursors is challenging (Rix et al., 2022). Due to their morphology, VSLSLCs can be mistaken for EVs and ABs in some assays and vice versa. The involvement of EVs and ABs in cellular development is an area of intense research, as many studies indicate (Denzer et al., 2000; Migneault et al., 2020; Phan et al., 2020; Beltraminelli et al., 2021; Gregory and Rimmer, 2023).
Very small cellular debris and EVs generally appear in scatter dot plot regions that are difficult to quantify. EVs and ABs may rarely mimic VSLSLCs, as they are a few micrometers in size and can contain strong condensed DNA fragments, similar to cell nuclei, surrounded by a double membrane with surface markers. They are differentiated from VSLSLCs by their round membranes, alkaline pH environment, and immature, less condensed chromatin. The 7-AAD dye aids in distinguishing them on scatter dot plots from entities with intact membranes. However, since some EVs and ABs are similar in size and content and can display CD markers, they may not allow the entry of 7-AAD and similar dyes in early stages, complicating their differentiation from (very) small cell developmental stages. We hypothesize that VSLSLCs function as precursors to LSLCs, and their numbers in cell density-dependent different leukemic sublines, along with other developmental stages, should exhibit significant differences (Lica et al., 2018; Lica et al., 2021). Considering the data regarding CD markers for VSELSCs, we propose that hematopoietic progenitor cell antigen CD34 could be one of the first markers observed on the cell surface. For the negative marker for VSELSCs, we selected receptor-type tyrosine-protein phosphatase C (CD45 antigen) since it is commonly used by researchers (Lacombe et al., 1997; Kuruca et al., 2019; Filidou et al., 2023) and is part of the EuroFlow standardization for hematological probes (Van Dongen et al., 2012; Glier et al., 2019). CD markers that exclusively target VSELSCs, such as NANOG and CD44, stage-specific embryonic antigen-4 (SSEA4), CD133 antigen, show lower efficiency in cell lines (Gunjal et al., 2015; Sellers et al., 2017) than in samples directly from organisms and CD44 antigen can be present on the surface of apoptotic bodies, potentially invalidating and distorting results by causing false positives or negatives. To ensure consistency in interpreting findings from previous studies (Lica et al., 2018; Lica et al., 2021) and to distinguish between different developmental stages within the tested cell lines, we chose neural cell adhesion molecule (CD56 antigen) as an indicator of a poorer prognosis in multiple myeloma (Lanier et al., 1989; Zheng et al., 2022). To confirm the presence of DNA and its degree of condensation, we examined the CD34 marker level relative to DNA content.
Briefly, VSLSLCs can directly increase their numbers and indirectly enhance the number of lower hierarchical developmental cell stages in culture. They are likely to exhibit a triple-negative status (CD45−/CD56−/CD2−) for the tested CD markers and a positive status for CD34, with morphology exhibiting alkaline cytoplasm and less condensed DNA compared to ABs while being negative for 7-AAD. However, cytological analysis indicates that the populations of the most primitive cells can be divided into CD34− VSLSLCs and CD34+ SLSLCs (Lica and Pradhan, 2023). Notably, CD34 is a surface protein commonly associated with HSCs and is used as a marker in various leukemia types, including acute lymphoblastic leukemia (ALL) and AML. However, studies show that a significant subset of patients with ALL and AML do not express CD34 on leukemic cells (Aljurf et al., 2011; Ho et al., 2020; Araki et al., 2023; Araki et al., 2024). Research has demonstrated that CD34− cells can still exhibit SC characteristics, especially among leukemic populations, indicating that CD34 is not essential for identifying or isolating LSCs in vitro (Lica and Pradhan, 2023).
CD34 is also not characteristic of mesodermally specified embryoid bodies that form HPCs (Bauwens et al., 2008; Demirci et al., 2020; Anjos-Afonso and Bonnet, 2023). Mesodermally specified embryoid bodies typically range in size from approximately 150–450 μm, depending on culture conditions and developmental stage. Their size significantly influences their differentiation potential, with smaller bodies favoring endodermal differentiation and larger ones supporting mesodermal development. Controlling their size is a crucial factor in optimizing differentiation protocols for specific lineages (Bauwens et al., 2008; Demirci et al., 2020).
Given the morphological similarities among these structures, we provide an overview of their distinct characteristics in Table 2.
For more detailed information, please refer to our previous work (Lica et al., 2018; Lica et al., 2021; Lica and Pradhan, 2023).
Cytological characteristics of small stem progenitors in embryos and adultsEpibCsIn humans, these cells typically measure 10–15 µm in size. While their size may vary slightly depending on the cell cycle phase, it generally falls within this range. At the epiblast stage, the cells are pluripotent, meaning they can develop into any cell type in the body, including those from the ectoderm, mesoderm, and endoderm (Figure 1). ESCs (EpibC precursors) appear at an early stage of development and are extracted from the ICM of the embryo during the blastocyst phase before the embryo implants in the uterus. The blastocyst forms around 5–6 days after fertilization (Figure 2); at this stage, the embryo consists of two main groups: the ICM, which will develop into the fetus, and the trophectoderm, which surrounds the ICM and contributes to forming structures like the placenta (Thomson et al., 1998).
MdPCsIn adult humans, these cells are similar in size to other types of progenitor SCs, typically measuring around 10–15 µm. Their size varies based on their developmental and differentiation stage (Asahara et al., 1997; Erices et al., 2000; Igura et al., 2004; Kögler et al., 2004; Zhang et al., 2011; Divya et al., 2012; Lyahyai et al., 2012; Slukvin and Kumar, 2018). As these cells specialize into specific cell types, such as muscle, bone, or blood cells, their dimensions may adjust to suit the requirements of their final function. They can differentiate into mesenchymal and endodermal lineages, although their pluripotency gene expression is limited (Figure 1). These cells exhibit regenerative potential, like VSELSCs, and express surface markers such as endoglin (CD105), 5′-nucleotidase (CD73), and Thy-1 membrane glycoprotein (CD90) but not CD45, aligning with their role in tissue regeneration (Asahara et al., 1997; Erices et al., 2000; Igura et al., 2004; Kögler et al., 2004; Zhang et al., 2011; Divya et al., 2012; Lyahyai et al., 2012; Slukvin and Kumar, 2018).
EnthPCsThese cells measure 10–15 μm, primarily differentiate into endothelial cells, and have limited potential for forming other cell types (Figure 1). They play a crucial role in vascular regeneration and share molecular similarities with VSELSCs, especially regarding their regenerative capabilities. EnthPCs are characterized by the expression of markers such as CD34, vascular endothelial growth factor receptor 2 (VEGFR2), CD133 antigen, and other markers that are associated with endothelial cells (Aprile et al., 2024).
MsPCsThese cells can differentiate into various specialized cells, including osteocytes (bone cells), chondrocytes (cartilage cells), and adipocytes (fat cells) (Figure 1). MsPCs can be isolated from diverse tissues, including bone marrow, adipose tissue, skeletal muscle, and umbilical cord tissue (Reyes et al., 2001; Kögler et al., 2004; Ulloa-Montoya et al., 2007; Kuroda et al., 2010; Phermthai et al., 2010; Demerdash et al., 2020; Chamling et al., 2021; Budeus et al., 2023; Ghabriel et al., 2023; Ebrahim et al., 2024). Typically, MsPCs measure around 15–30 µm in size, although their dimensions can change based on the culture environment, and reports indicate that they may shrink to as small as 6–8 µm (Han et al., 2019; Qu et al., 2024). The key molecular signaling pathways for MsPCs include BMP, which is essential for the differentiation into osteocytes and chondrocytes; Wnt/β-catenin, which regulates cell proliferation and differentiation; NOTCH, which is important for SD and differentiation, particularly towards muscle cell formation; the phosphatidylinositol 3-kinase (PI3K)/Akt which governs cell survival, proliferation, and differentiation; and Hippo-transcriptional co-activators Yes-associated protein (YAP)/transcriptional co-activator with PDZ-binding motif (TAZ), which regulates cell growth, differentiation, and response to mechanical stress. MsPCs are characterized by specific positive surface markers such as CD105+, CD73+, CD90+, and CD44+ while lacking hematopoietic markers CD11b−, CD14−, CD19−, CD34−, CD45−, CD79a−, or HLA class II histocompatibility antigen, DR alpha chain (HLA-DR−) and pluripotency markers such as OCT4 and SSEA4 (Pittenger et al., 1999; Caplan, 2005; Dominici et al., 2006; Phinney and Prockop, 2007; Uccelli et al., 2008).
Long-term HSCs (LT-HSCs)LT-HSCs are small, comparable in size to small lymphocytes, with a typical diameter of 7–10 µm (Figure 1). They have the capacity for long-term SD and can function throughout an organism’s lifetime. LT-HSCs maintain a distinct epigenetic program characterized by a closed chromatin state, which preserves gene silencing and prevents unwanted differentiation. Their low metabolic activity and slow cell cycle mean they can sustain their SD ability for extended periods (Wang and Ema, 2016). These cells are quiescent, entering dormancy to protect against environmental stresses, including oxidative damage. They rely on glycolytic metabolism, which lowers oxygen consumption and minimizes oxidative stress. LT-HSCs express genes such as forkhead box O1 (FOXO1), transforming growth factor beta 1 (TGFB1) and NFE2 like bZIP transcription factor 2 (NFE2L2), which are involved in maintaining quiescence and defending against oxidative stress. Autophagy plays a critical role in clearing damaged organelles. Compared to short-term HSCs (ST-HSCs), LT-HSCs exhibit higher activity in stress-protective pathways, including TGFβ, p53, and FoxO, which enhance their long-term SD potential. The Wnt pathway is also more active in LT-HSCs, helping regulate their regenerative capacity. Additionally, LT-HSCs exhibit a more closed chromatin state, further preserving gene silencing and preventing premature differentiation. Their higher expression of oxidative stress protection genes equips LT-HSCs to tolerate damage that could lead to cellular degeneration. Conversely, ST-HSCs are more susceptible to damage but are more efficient in short-term blood cell production (Wilson et al., 2007). Notably, human LT-HSCs and ST-HSCs express slightly different surface markers than their mouse counterparts, particularly regarding CD34; human LT-HSCs are CD34+, while mouse LT-HSCs are CD34−. Other markers, such as CD38, CD90, CD45RA, and CD49f, help distinguish LT-HSCs from ST-HSCs. Interestingly, certain conditions may give rise to a CD34− LT-HSC population in humans, which may support long-term renewal and multipotency (Notta et al., 2011; Vedi et al., 2016; Ho et al., 2020).
ST-HSCsST-HSCs vary in size depending on their cell cycle stage, with diameters ranging from 8 to 12 µm. They have limited SD capacity and are more metabolically active than LT-HSCs. ST-HSCs primarily participate in the rapid replenishment of blood cells and are crucial for short-term regenerative functions. Initially believed to support hematopoiesis for only 4–12 weeks, recent studies indicate their capacity may extend beyond this period. Experimental models have shown ST-HSCs can maintain engraftment for up to 1 year in primary recipients and at least 3 months in secondary recipients, suggesting a broader long-term potential than previously assumed; however, their overall engraftment ability remains lower than that of LT-HSCs. Despite their restricted SD ability, ST-HSCs predominantly differentiate HSCs/multipotent progenitor cells (MPPs), rapidly replenishing blood cell populations. They enter the cell cycle more quickly than LT-HSCs, facilitating faster differentiation. Key signaling pathways such as Janus kinase (JAK) signal transducer and activator of transcription (JAK-STAT) (promoting proliferation and differentiation), mammalian target of rapamycin (mTOR) (whose excessive activation can deplete SD), and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) (MAPK/ERK) (involved in proliferation and differentiation) are more active in ST-HSCs. Compared to LT-HSCs, ST-HSCs have a more “open” chromatin state, increasing transcriptional accessibility to genes related to differentiation and proliferation. Genetictened mitotic activity renders them more susceptible to genetic damage and oxidative stress, particularly from reactive oxygen species, due to their reliance on oxidative phosphorylation. The increased activity of proliferation and differentiation pathways drives ST-HSCs to differentiate more rapidly, though this acceleration limits their regenerative potential. Unlike LT-HSCs, ST-HSCs express surface markers such as CD34 and CD38 but not CD90, correlating with their rapid differentiation capacity (Wilson et al., 2007; Cabezas-Wallscheid et al., 2014; Zeng et al., 2022).
Other primitive adult PSCsSeveral additional PSCs in the adult human body are similar in size, regenerative capacity, and differentiation potential to VSELSCs (Bhartiya et al., 2022).
Marrow-isolated adult multilineage inducible cells (MIAMICs)These cells are a unique population derived from human bone marrow, typically 6–12 µm in size (D’Ippolito et al., 2004) (Figure 1). They are a specialized subset of MsPCs that, under appropriate in vitro conditions, can differentiate into various cell types, including osteoblasts, chondrocytes, adipocytes, and even nerve cells. MIAMICs express key pluripotency genes, such as POU5F1P5 (synonym OCT4), SOX2, and NANOG, albeit at lower levels than ESCs or iPSCs. Notably, MIAMICs can survive in challenging environments, including hypoxia, indicating a resilience not commonly seen in other adult SCs. Their expression of SSEA4 and other pluripotency genes, coupled with their stress resistance, suggests they may have enhanced flexibility in differentiation. MIAMICs are characterized by the absence of hematopoietic markers (CD34− CD45−) and the presence of CD29+, CD73+, CD90+, CD105+, and SSEA4+, a marker used to identify pluripotent cells, including ESCs (D’Ippolito et al., 2004; Rossi et al., 2019).
Multilineage-differentiating stress enduring cells (MUSECs)MUSECs are SCs isolated from human bone marrow and various other tissues, generally measuring 10–15 µm in diameter (Kögler et al., 2004; Wakao et al., 2011) (Figure 1). These cells can differentiate into multiple lineages derived from all three germ layers: mesoderm, ectoderm, and endoderm. Their differentiation potential is linked to the expression of key pluripotency genes (Wakao et al., 2011, 2012; Leng et al., 2019). MUSECs express surface markers such as stage-specific embryonic antigen 3 (SSEA3), commonly found in PLSCs, and CD105, which is also a marker for MsPCs. However, they do not express CD90, CD34, or CD45. Their unique molecular characteristics, including the expression of pluripotency genes (OCT3, OCT4, SOX2, NANOG) and their ability to withstand stress and retain genomic stability make them promising candidates for regenerative therapies (Wakao et al., 2011).
CD133+ PSCsCD133+ PSCs have been identified in various human adult tissues, including peripheral blood, bone marrow, the brain, and even in cancerous tissues (Shmelkov et al., 2008; Wu and Wu, 2009; Kreso and Dick, 2014; Shaikh et al., 2015; Glumac and LeBeau, 2018; Wang et al., 2020; Korn et al., 2021; Nayak et al., 2022; Liu et al., 2023). Their median diameter is approximately 5–10 μm, typical for small, primitive hematopoietic cells (Figure 1). This size variation may indicate adaptation to specific microenvironments and potentially reflect a more primitive or specialized function within the hematopoietic system (Hawley et al., 2006; Mizrak et al., 2008). CD133+ PSCs express markers characteristic of various PSC types, such as those of endothelial, hematopoietic, and neurogenic origins. However, unlike VSELSCs, CD133+ PSCs express lower levels of pluripotency markers, indicating a more specialized state. Importantly, CD133 is not exclusive to SCs; it is also found in various types of healthy cells and MCs (Kreso and Dick, 2014; Korn et al., 2021; Liu and Qian, 2021; Nayak et al., 2022). CD133 has gained attention as a CSC marker in cancer research, suggesting its involvement in tumor formation and resistance to cancer therapies. The potential use of CD133+ PSCs in regenerative medicine, particularly for heart tissue repair, vascular regeneration, and neurogenesis, is an area of growing interest. However, the role of CD133 as a definitive marker for precursor and progenitor SCs remains debated.
Node and duct SCs (NDSCs)NDSCs are SCs derived from nodes and ducts within the body, primarily associated with hematopoietic and neural systems (Lee et al., 2014). Present in the vascular systems, these cells can differentiate into neuronal cells in vitro. In an in vivo model of ischemic brain injury, NDSCs exhibited promising regenerative effects, highlighting their proliferative potential (Lee et al., 2014). Typically ranging from 6 to 8 µm in diameter, NDSCs share characteristics with both HSCs and neural SCs, given their ability to differentiate into both blood and neuronal cells. They are relatively small, with sizes comparable to LT-HSCs (Figure 1). Their small size enables them to effectively circulate through the vascular system, which is crucial for their role in tissue repair and regeneration. NDSCs are regulated by several key molecular pathways during differentiation, including the BMP, fibroblast growth factor (FGF), and retinoic acid (RA). They exhibit a distinct immunophenotype, often expressing key surface markers such as CD34 (commonly used to identify SCs, particularly HSCs and MsPCs), CD73 (indicating immunomodulatory capacity and support for regenerative processes), CD90+ (associated with tissue repair and regenerative potential), and CD44 (involved in cell adhesion to the extracellular matrix and SC mobility, playing a role in tissue repair) but not Lin (lineage negative, indicating the absence of markers typical of mature, committed cells) and CD45 (characteristic of mature immune and blood cells). These markers are essential for identifying and isolating NDSCs from other SC types. NDSCs activate several signaling pathways that regulate their proliferation, differentiation, and responses to environmental cues: Wnt/β-catenin regulates SD and enhances proliferation; NOTCH controls differentiation and helps maintain the SC state; TGF-β plays a role in maintaining SC homeostasis by regulating the transition from quiescence to active differentiation and influencing the immunomodulatory abilities of these cells; PI3K/AKT/mTOR controls proliferation and survival (mTOR overactivation may reduce regenerative capacity, but modulating this pathway supports their repair functions); and hypoxia-inducible factors (HIF) facilitates adaptation and function in low-oxygen niches, promoting survival and maintaining pluripotency in stressful, oxidative conditions. These pathways guide the cells through processes that lead to specific lineage commitments depending on environmental cues and specific growth factors. Research into the therapeutic application of NDSCs has revealed their potential for treating neurological disorders, autoimmune diseases, and cancer by targeting damaged tissues and supporting cellular regeneration (Lee et al., 2014
留言 (0)