In this pre-specified analysis, the principal finding is that persistent myocardial ischaemia is infrequent in patients recently hospitalised with COVID-19 and elevated cardiac troponin. This finding coupled with the lack of an association between ischaemia and myocardial scar suggests that epicardial coronary artery disease (CAD) is unlikely to be the predominant mechanism underlying COVID-19 induced myocardial injury. Rather, it is more likely that myocardial injury in COVID-19 is thrombotic or thromboembolic in origin and hence, standard therapies for acute coronary syndromes may not be warranted in the majority of patients with troponin elevation in COVID-19.
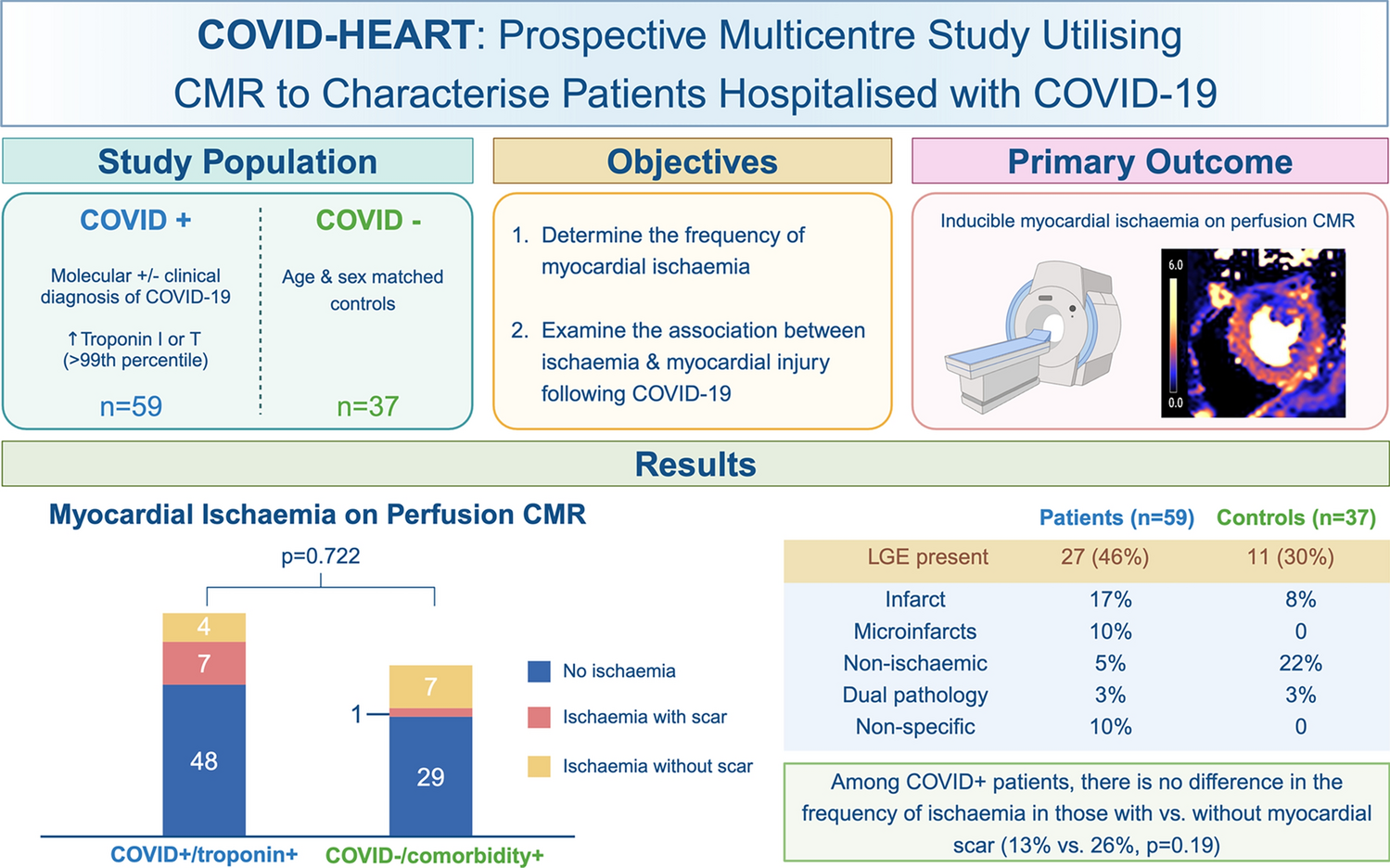
Early studies of acute cardiac injury following COVID-19 revealed abnormalities in up to 78% of patients in some series [11, 23, 24]. However, the prevalence of cardiac pathology attributable to COVID-19 may have been overestimated in small and/or single-centre studies which adopted a retrospective approach and lacked appropriate contemporaneous matched controls. In COVID-HEART, a multicentre, prospective study with contemporaneous matched controls, cardiac abnormalities were observed in 61% of COVID + /troponin + patients, twice the prevalence of that in either COVID + /troponin− subjects (36%) or COVID − /comorbidity + controls (31%) [15]. Infarction and microinfarction were more prevalent (respectively, 13 vs. 2 vs. 7%, and 9 vs. 0 vs. 1%, p < 0.01), but non-ischaemic scar was comparable (13 vs. 5 vs. 14% p = N.S.). Several explanations are plausible, including a higher prevalence of pre-morbid coronary heart disease in individuals with myocardial injury complicating COVID-19 and second, the vascular pathophysiology of acute COVID-19, including macro- and microangiopathic thrombosis contributing to ischaemic myocardial injury following COVID-19 [25].
Acute cardiac injury in COVID-19 is thought to involve a complex interplay of factors, including direct viral cytopathic effects, immune response dysregulation and dysfunction of the renin–angiotensin–aldosterone system (RAAS) [26, 27]. Although angiotensin-converting enzyme 2 (ACE2) receptor-mediated viral entry and RAAS dysfunction is unique to COVID-19, the damage caused by the excessive activation of the innate immune system and release of pro-inflammatory cytokines is common to sepsis from other aetiologies. These mechanisms may give rise to structural and/or functional abnormalities of the microvasculature, resulting in impaired perfusion and consequent cell damage [28]. In the myocardium, viral interaction with ACE2 receptors expressed on capillary pericytes may increase levels of angiotensin II, a powerful vasoconstrictor which may contribute to vascular injury, as well as augmenting inflammation and thrombogenicity [29,30,31]. Thus, de novo inflammatory damage to the endothelium and/or augmented inflammatory activity within existing atherosclerotic plaques may ensue. A key feature of COVID-19 is hypercoagulability, mediated by increased platelet activation and degranulation, increased neutrophil extracellular traps and activation of the contact/tissue factor pathways [32, 33]. The resultant microthrombi deposition accounts for the high incidence (25%) of venous thromboembolism and thrombotic events in multiple organ systems [5,6,7,8]. Accordingly, elevated levels of D-dimer/fibrinogen are associated with adverse outcomes [34]. Hence, both inflammatory and thrombogenic mechanisms, culminating in obstruction of coronary arteries, may underlie COVID-19-associated damage within the heart. Consistent with this, autopsy studies have confirmed the presence of microthrombi within the coronary microvasculature [35, 36].
Although several CMR studies have investigated myocardial injury, relatively few have evaluated myocardial perfusion following COVID-19. A single-centre retrospective study of patients referred clinically for investigation of chest pain and/or dyspnoea with myocardial perfusion scintigraphy by single-photon emission computed tomography (MPS-SPECT) identified a higher prevalence of ischaemia in patients with previous COVID-19 (23%; 77/329) than in those without (16%; 244/1495) [9]. In a case–control study of 34 post-COVID-19 subjects referred for clinically indicated N13-ammonia myocardial stress perfusion PET (studied a median time of 4.6 months after COVID-19), abnormal myocardial blood flow reserve (< 2) was identified in 44% (15/34) of subjects compared with 12% (12/103) of matched, COVID-19 negative controls (p < 0.001) [13]. Reduced MPR was also demonstrated in a small single-centre study evaluating coronary sinus flow using CMR in post-COVID-19 patients (n = 22, studied 1–6 months after acute illness) [10]. However, this study was restricted to patients with persistent post-COVID-19 dyspnoea and fatigue, and controls (n = 17) were selected retrospectively, with low pretest probability of CAD and were significantly younger (median age 39 years versus 51 years for patients). Another study evaluated 148 patients hospitalised with severe COVID-19 (with troponin elevation, and 32% requiring ventilatory support) [11]. Ischaemia testing with CMR was carried out when a clinical indication was present (n = 76, median 68 days following discharge): inducible ischaemia was present in 26% (20/76). In a case–control study of 90 post-COVID patients clinically referred for adenosine stress-perfusion CMR, regional perfusion defects were identified in 36% (32/90) of patients [12]. On quantitative flow assessment, compared with 90 age-, sex- and comorbidity-matched controls (studied pre-COVID-19), there was no significant difference in global hyperaemic MBF. The authors concluded that the high prevalence of regional ischaemia and/or infarction (40%) suggested the likely presence of pre-existing occult CAD, reflecting the demographics and comorbidity burden of the population studied (elderly with multiple cardiovascular risk factors).
Limitations inherent in previous studies include selection bias from the study of patients referred clinically for ischaemia assessment and the use of convenience samples of patients presenting with chest pain or dyspnoea (some studies excluding those with prior CAD). The majority have also been retrospective analyses, with a wide interval between the index admission and perfusion assessment. Another limitation is the lack of appropriate contemporaneous, matched controls. Nonetheless, these data are largely consistent with our findings, taken from a prospective study with matched controls, that myocardial ischaemia is not significantly more prevalent than in a matched control population.
Subjects studied in our analysis were imaged a median of 21 days after hospital discharge. Although this is earlier than in all previously published studies, it is still possible that pro-inflammatory changes and hypercoagulability may be transient phenomena which may have resolved either spontaneously or with medical therapy (with antiplatelet/anticoagulant agents) prior to CMR assessment. Hence, the pathophysiological changes responsible for myocardial damage may not have been captured at the time of CMR assessment, and the extent of hypoperfusion in the acute phase, underestimated. There is some evidence that microvascular dysfunction in COVID-19 may be regional, in contrast to the global microvascular dysfunction that is observed in non-obstructive CAD or in association with type 2 diabetes. This may reflect pathophysiological differences in the structural and functional elements of disease, with transient thrombosis and inflammation being more prominent in COVID-19.
An interesting observation is that a significant proportion of patients with myocardial injury did not have stress perfusion defects. A number of explanations may apply. Firstly, stress perfusion imaging may lack sensitivity relative to LGE imaging for myocardial injury, owing to its lower spatial resolution and its imaging being confined to three short-axis slices (compared with full left ventricular coverage with LGE imaging). Secondly, microvascular abnormalities may also be regional, corresponding to the microinfarcts observed in 9% of cases. Even quantitative flow assessment may lack sensitivity in identifying these regional differences, though a pixel-wise approach, as used in the majority of our participants, is a step forward. Thirdly, it is possible that troponin elevation may relate not only to macro- or microvascular disease mechanisms, but also to myocardial inflammation/myocarditis including direct viral cytopathic effects, or type 2 myocardial infarction (due to oxygen supply–demand imbalance). Another potential injury mechanism in COVID-19, which may underlie discordance between scarring and hypoperfusion, is the occurrence of tissue ischaemia with normal [or even high] myocardial blood flow. However, the observed patterns of injury (namely predominantly macro and/or microinfarcts) indicate that damage may be mediated by vascular mechanisms, though this may be due to a transient prothrombotic state.
Study limitations
A limitation of our study is that only a subset of participants in COVID-HEART underwent stress perfusion imaging, with the latter being determined according to the availability of stress perfusion CMR in the recruiting centre and physician/patient choice. Other potential sources of bias include the exclusion of patients with contraindications to CMR and the exclusive recruitment of subjects with biochemical evidence of myocardial injury: hence, the results of this work may not be representative of wider COVID-19 population (namely those without overt cardiac injury, and non-hospitalised patients, in whom ischaemia may still occur). Nonetheless, this remains a real-world multicentre clinical cohort recruited prospectively and compared with contemporaneous controls analysed in a blinded core lab. However, survivor bias remains another possible limitation: more severe injury and microvascular obstruction may occur in more severe/end-stage disease (as indicated in some autopsy series). Without anatomical assessment (invasive angiography or computed tomography coronary angiography), the prevalence of epicardial CAD could not be determined, and without invasive assessment, CMR-determined microvascular dysfunction could not be definitively confirmed as such (as opposed to multivessel CAD).



留言 (0)