
記住我

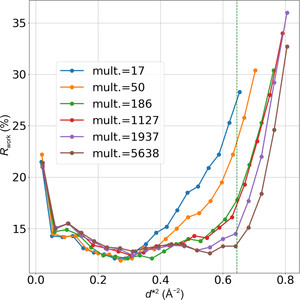
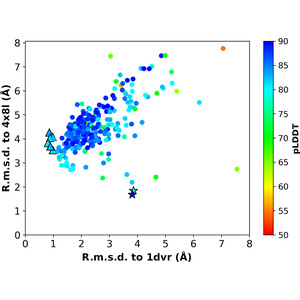




Molecular replacement (MR) is highly effective for biomolecular crystal structure determination, increasingly so as the database of known structures has increased. For candidates without recognizable similarity to known structures, however, crystal structure analyses have nearly always required experiments for de novo phase evaluation. Now, with the unprecedented accuracy of AlphaFold predictions of protein structures from amino-acid sequences, an appreciable expansion of the reach of MR for proteins is realized. Here, we sought to automate an AlphaFold-guided MR procedure that tailors predictions to the MR problem at hand. We first optimized the reliability cutoff parameters for residue inclusion as tested in application to a previously MR-intractable problem. We then examined cases where AlphaFold by default predicts a conformation alternative to that of the candidate structure, devising tests for MR solution either from domain-specific predictions or from predictions based on diverse sequence subclusters. We tested subclustering procedures on an enzyme system that entails multiple MR-challenging conformations. The overall process as implemented in Phenix automatically surveys a succession of trials of increasing computational complexity until an MR solution is found or the options are exhausted. Validated MR solutions were found for 92% of one set of 158 challenging problems from the PDB and 93% of those from a second set of 215 challenges. Thus, many crystal structure analyses that previously required experimental phase evaluation can now be solved by AlphaFold-guided MR. In effect, this and related MR approaches are de novo phasing methods.
留言 (0)