
記住我

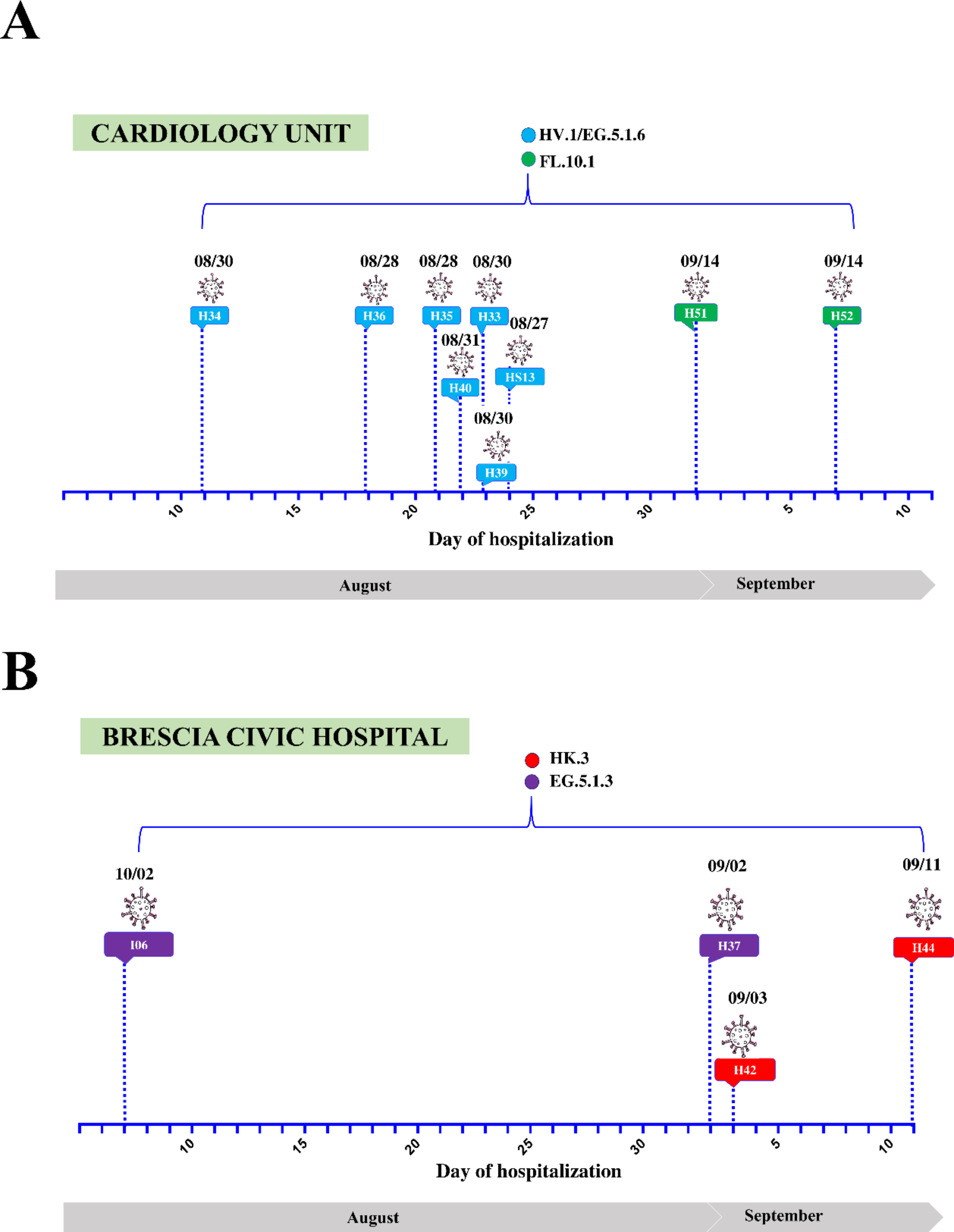

Following the onset of respiratory symptoms, nine patients (HS13, H33, H34, H35, H36, H39, H40, H51, and H52) who were hospitalized in the cardiology unit of the Brescia Civic Hospital between late August and September 2023, tested positive for SARS-CoV-2 (Fig. 1A). Since all patients were negative for SARS-CoV-2 infection based on molecular diagnostic testing at the time of admission, it is likely that they acquired the virus during hospitalization. In order to define the root source of the outbreak, whole genome sequencing (WGS) analysis was performed on nasopharyngeal specimens. In particular, WGS revealed that patients were infected with the following Omicron sub-lineages: HV.1 (H33, H34, H35, H36, H39, H40), EG.5.1.6 (HS13), and FL.10.1 (H51, H52) (Fig. 1A).
To establish whether the cases found in the cardiology ward represent a specific and confined nosocomial outbreak, patients (H37, H42, H44, I06) hospitalized in four different hospital’s units (emergency medicine unit, emergency room, general medicine unit and neuropsychiatry unit) were also included in the study as a control group. Differently from the patients belonging to the cardiology ward, they were infected with Omicron sub-lineages belonging to HK.3 (H42, H44), and EG.5.1.3 (H37, I06) lineages (Fig. 1B).
Fig. 1
Timeline of hospitalization and SARS-CoV-2 positivity of patients’ hospitalized in the (A) cardiology unit or (B) other units of Brescia Civic Hospital. SARS-CoV-2 lineages are highlighted in different colors. indicates the day when each patient tested positive for SARS-CoV-2 infection
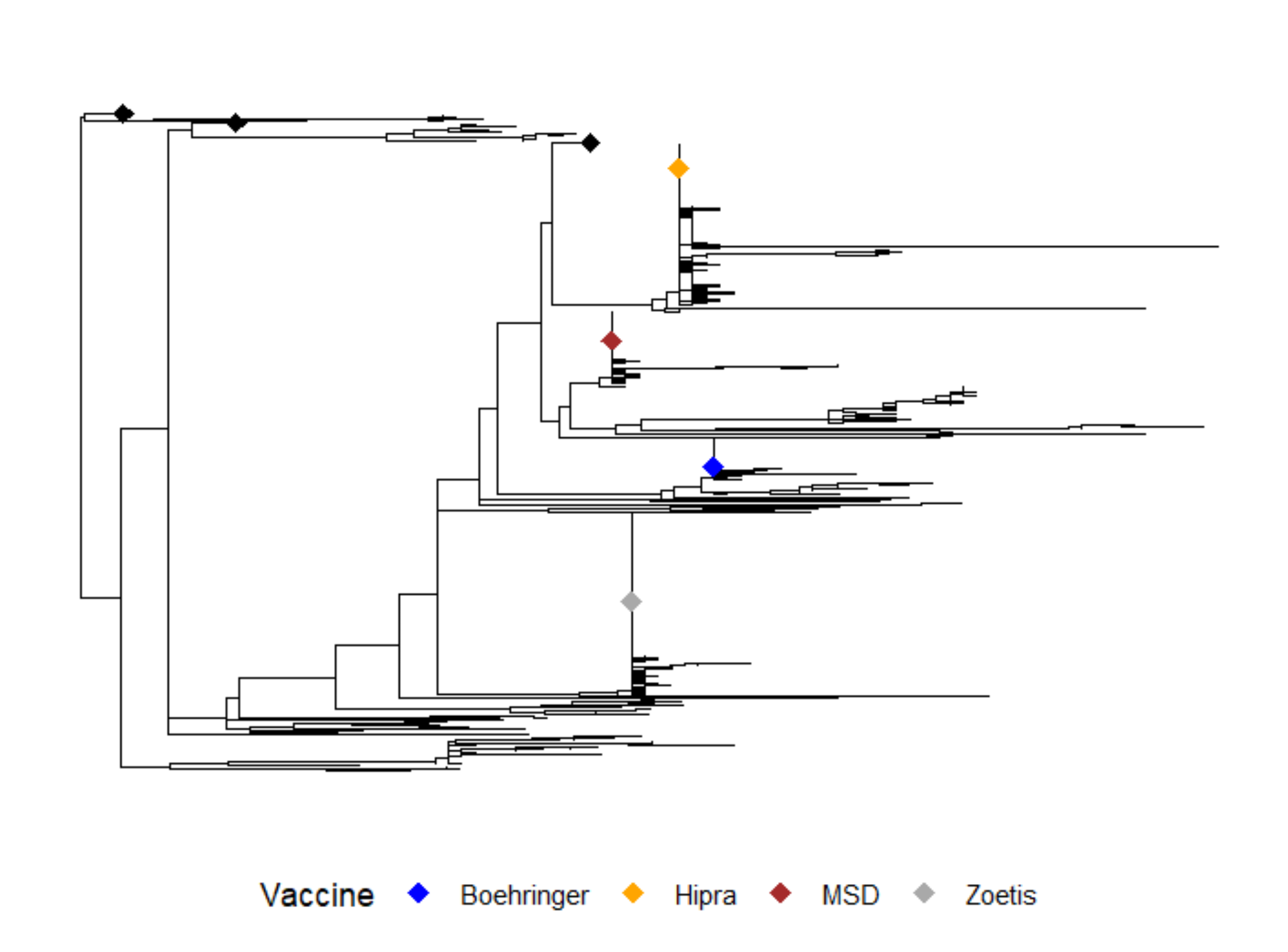
Epidemiological analysisTo clarify the transmission events leading to the outbreak and to understand if there was a common source of infection, we inferred a time-stamped phylogenetic tree placing samples collected from the cardiology unit within a larger context of SARS-CoV-2 genetic variations. In addition to our cases, representative sequences from Omicron variants circulating in Italy between November 2022 and October 2023 were evaluated. As shown in Fig. 2, the analysis revealed that consensus sequences collected from patients HS13, H33, H34, H35, H36, H39, and H40 sited close together on the phylogenetic tree (Cluster 1), while patients H51 and H52 formed a small distinct cluster (Cluster 2). Of note, the consensus sequence of patient HS13, in spite of belonging to the cardiology HV.1 group (Cluster 1), tested positive for a distinct SARS-CoV-2 Omicron variant (EG.5.1.6) and was located on a different branch of the tree. On the other hand, the control group patients H37, H42, H44, and I06 (yellow circles) were scattered across the phylogenetic tree (Fig. 2).
Fig. 2
Time-resolved maximum-likelihood tree of SARS-CoV-2 including 13 whole genomes obtained from the hospitalized patients considered for this study in addition with a set of representative Italian data (n = 2331) collected between November 2022 and October 2023. The genomes are colored according to the SARS-CoV-2 lineages. In addition, the Clusters 1 and 2, that include patients admitted to the cardiology unit identified in this study, are highlighted
Due to substantial sequence similarity among SARS-CoV-2 Omicron variants, the phylogenetic tree obtained with the consensus sequences, could not accurately determine the virus transmission chain among patients and, so, could not clearly solve the hospital outbreak.
Definition of virus transmission through bottleneck size evaluationEstimating the number of transmitted viral particles is crucial to track epidemiologically linked SARS-CoV‐2 infections [7]. The Beta Binomial inference method was used to calculate the bottleneck sizes and, together with the dates of hospitalization and samples collection, was taken into account in order to determine the transmission directionality among the SARS-CoV-2 positive patients. We performed 106 bottleneck size estimations, pairing randomly the patients and testing each patient both as a donor and recipient. H34, as shown in Fig. 1A, was the first patient admitted to the cardiology unit, so we speculated that the outbreak may have started with this patient. Then, each hospitalized subject was evaluated as donor or recipient in a hypothetical transmission chain according to the bottleneck dimension. This analysis (Fig. 3A) clearly showed the directionality of the viral transmission for some pairs, for example for H34-HS13, H34-H35, H34-H36, H39-H33, H40-H33, H39-H36, and H52-H51, while other subjects (H34, H39, H40), if coupled, displayed similar results in both directions, making it difficult to establish who is the donor and who is the recipient. Taking into consideration both bottleneck sizes and date of hospital admission, we reconstructed a hypothetical chain of viral transmission among the cardiology unit patients (Fig. 3B). In particular, a wide bottleneck was found when H34 was coupled with all the other patients from Cluster 1, supporting our hypothesis that this patient was the cluster’s index case. However, considering the bottleneck dimension, we hypothesized that patient H33 most likely acquired the infection via H39 (H39-H33 range: 53–200 Nb). Determining the H34-H39-H40 transmission chain was a challenging issue, due to a wide bottleneck for all the possible donor-recipient pairs (H34-H39 range: 20–200 Nb vs. H39-H34 range: 11–187 Nb; H34-H40 range: 33–200 Nb vs. H40-H34 range: 21–200 Nb, H40-H39 range: 60–200 Nb vs. H39-H40 range: 54–200 Nb). Although we do not exclude a possible transmission between H39 and H40, considering both the bottleneck dimension and the date of hospitalization/positivity, it is more likely that they both contracted the virus from subject H34. Concerning Cluster 2, both H51 and H52 tested positive for SARS-CoV-2 on September 14th. However, the bottleneck analysis suggests that H52 was the donor and H51 the recipient (H52-H51 range: 12–114 Nb vs. H51-H52 range: 2–4 Nb). Furthermore, a narrow bottleneck dimension was found with control patients (H37, H42, H44, I06), confirming the hypothesis that the SARS-CoV-2 cases detected in the cardiology department represent a confined nosocomial outbreak.
Fig. 3
(A) Maximum likelihood estimation for the mean transmission bottleneck size among paired-patients. Values represented as colored dots correspond to the hypothesized linked pairs, while the gray dots correspond to pairs that include at least one control patient. (B) Schematic representation of patients’ interaction across the epidemiological cluster. Arrows indicate the hypothesis of transmission direction. The subjects in the study are colored according to the SARS-CoV-2 lineages
Intrahost single nucleotide variants characterization and cluster analysis for genomic tracingTo further confirm the presence of two distinct clusters among the hospitalized patients in the cardiology unit, the Euclidean clustering method was performed to analyze the iSNVs found in the SARS-CoV-2 genome, as previously described [7]. As shown in Fig. 4A, the variations of iSNVs in terms of frequency and distribution along the whole genome displayed useful patterns to comprehend the viral transmission chains among patients. Data plotted in the heatmap (Fig. 4A) highlighted the close position of patients HS13, H33, H34, H35, H36, H39, H40 in the central branch and the localization of patients H51 and H52 in the same left branch, confirming the presence in the cardiology unit of the Cluster 1 and 2. Furthermore, Fig. 4A documents how patients I06, H37, H42, and H44, admitted to Brescia Civil Hospital in the same period but in different wards, were placed on distinct branches, separated from the clusters identified in the cardiology unit.
In order to validate our method, which considers the quasispecies found in a small portion of the SARS-CoV-2 genome, we conducted the same analysis taking into account the frequency and distribution of the iSNVs found in the nsp2, ORF3, and ORF7 genes only [7]. The Pearson correlation coefficients were used to evaluate the iSNVs variations and the results were plotted in a heatmap (Fig. 4B). As obtained with the whole genome, the iSNVs found in these 3 well conserved SARS-CoV-2 genes correctly identify transmission chains for both Cluster 1 and Cluster 2. In details, patients HS13, H33, H34, H35, H36, H39, H40 (Cluster 1) were plotted together in the central portion of the heatmap, while H51 and H52 formed a small distinct cluster (Cluster 2) in the left portion. Once again, our analysis confirmed that the iSNVs found in these genes can be utilized as a peculiar signature for contact tracing when the consensus sequences are identical.
Fig. 4
(A) Cluster heatmap built up with the iSNVs found in the whole SARS-CoV-2 genome, based on the similarity between samples. (B) Cluster heatmap constructed with iSNVs identified in the nsp2, ORF3, and ORF7 genes. Colors are defined according to the scale
留言 (0)